在上期的“走进科学,走进基因突变”一文中,小医带大家了解了基因突变的简单知识,相信大家对基因突变都有了一定的认知。基因突变作为当今生命科学的研究热点之一,检测方法也随之迅速发展,检测基因突变能够促进疾病的早期诊断和治疗。下面简述几种常用的基因突变检测方法及其基本原理,可根据检测目的和实验条件选择时参考。
一、Southern blot技术
Southern印迹杂交(Southern blot)是1975年由英国生物学家Edwin Southern发明,为了纪念该技术的诞生将其命名为Southern blot。该技术是最早出现的blot技术,是分子生物学领域中最常用的检测DNA的方法之一。其基本原理是具有一定同源性的两条核酸单链在一定条件下,可按碱基互补原则特异性地杂交形成双链。如果待检物中含有与探针互补的序列,则二者通过碱基互补的原理进行结合,游离探针洗涤后用自显影或其它合适的技术进行检测,从而显示出待检的片段及其相对大小(图1)。因此,该技术一经推出后便成为探针杂交领域最为经典的分子检测方法,被广泛应用于各种基因突变及限制性酶切片段长度多态性(Restriction Fragment Length Polymorphism,RFLP)的鉴定中。更多Southern blot详情,参见“走近实验技术中的“四大发明”之Southern blot、Northern blot和Western blot”一文。

图1 Southern Blot流程示意图(图片源自:National Human Genome Research Institute)。
二、PCR技术
对于基因突变的检测,1985年以前,大都采用Southern blot,其可筛选出基因缺失、插入和移码重组等多种突变形式。由于当时尚无法对样本中的靶基因进行人为扩增,对于那些Southern blot检测不到的突变,就只能通过诱变引物建立体外寡核苷酸介导的DNA突变进行复杂的DNA序列测定分析来确定,但该方法不仅复杂,还费时费力,并且成本较高。随着聚合酶链式反应(PCR)技术的兴起,目前几乎所有基因突变检测的分子诊断技术都建立在PCR基础之上,其自动化程度以及分析时间、结果准确性都得到了进一步提升,PCR技术的出现是基因突变检测研究的重大进展。下面小医就分别介绍几种临床上常用的基因突变检测技术的原理吧。1、ARMS-PCR
扩增阻滞突变系统PCR(Amplification Refractory Mutation System PCR,ARMS-PCR),又称等位基因特异性PCR(Allele-Specific PCR,AS-PCR),其建立在等位基因特异性延伸反应基础上,只有当某个等位基因特异性引物的3’端与突变位点发生碱基互补时,才能进行延伸反应。ARMS-PCR已成为国际上肿瘤个体化分子检测最重要、最先进的技术之一,同时也是遗传病诊断最准确的技术工具之一。
ARMS-PCR与普通PCR的区别在于引物设计时等位基因两条上游引物的3’端序列不同,当进行PCR扩增时,与模板不能完全匹配的上游引物将不能与模板完全形成互补碱基对,形成错配,当错配达到一定程度时,引物延伸将终止,得不到特异长度的PCR扩增产物,从而提示模板DNA没有与引物3’端配对的碱基,反之亦然(Peng et al.2018)。目前,科学家们已在ARMS-PCR方法基础上延伸出了四引物(Tetra-Primer ARMS-PCR),即同时设计四条PCR引物,其中两条位于3’端SNP位点上,并且方向相反且分属不同基因型,另外两条位于SNP外侧(图2)。
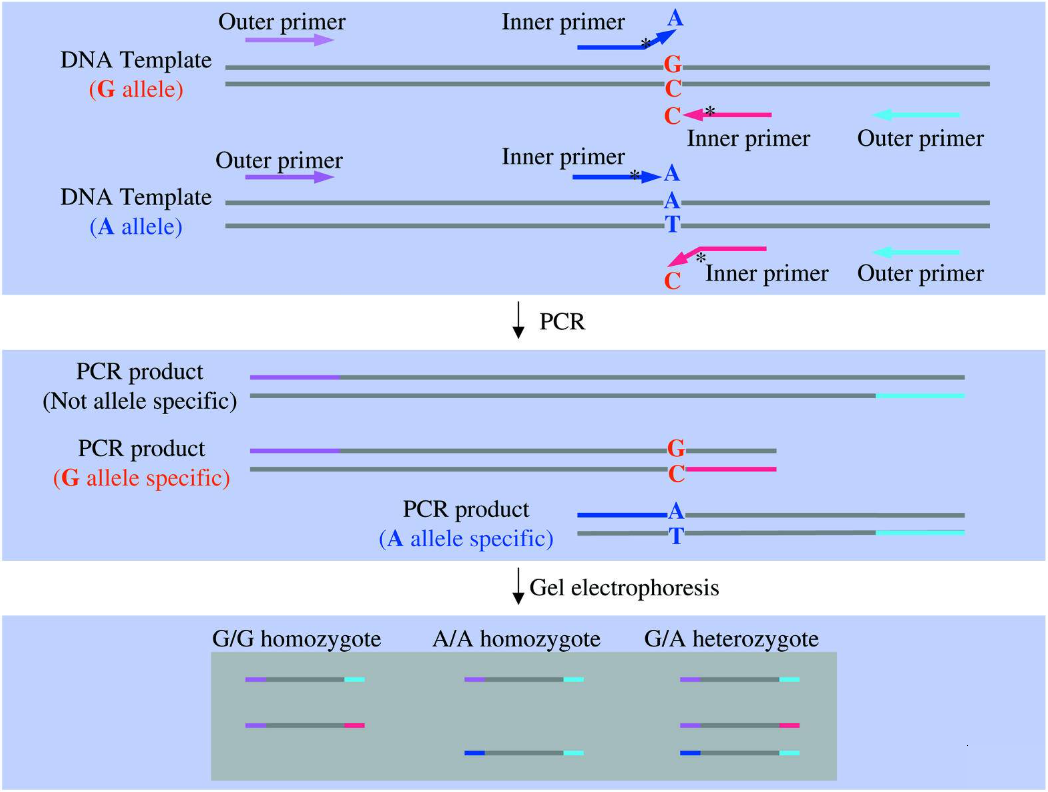
图2 四引物ARMS-PCR方法的示意图(Ye et al., 2001)。不同颜色表示参与PCR反应的不同引物。2、数字PCR
数字PCR(Digital PCR,dPCR)是一种核酸分子绝对定量技术,即第三代PCR技术,是生命科学和医学诊断的关键技术之一。1999年,Bert Vogelstein和Kenneth W.Kin-zler正式提出了数字PCR的概念。其通过将待测样本进行微滴化处理,使每个反应室中平均只含有一个或没有目标DNA分子,当液滴中的靶点被PCR扩增后,扩增结束后对各个反应单元的荧光信号进行统计学分析,有荧光信号记为1,无荧光信号记为0,得到检测样本中突变型和野生型的绝对定量,可检测到低至一个拷贝的突变(图3)。
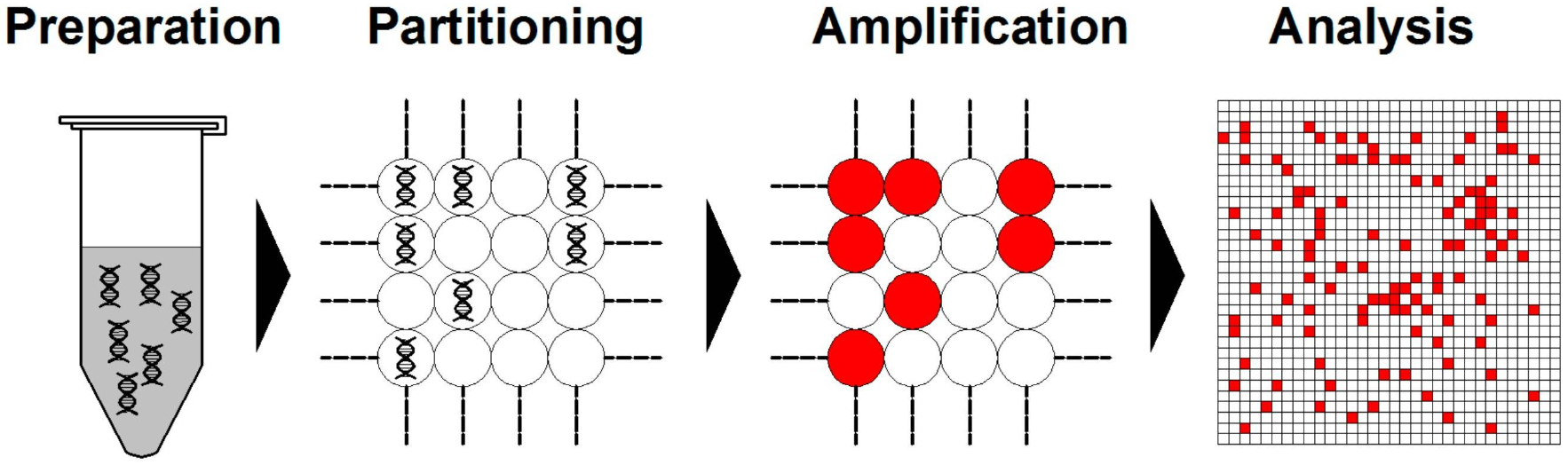
图3 典型数字PCR工作流程方案(Cao et al., 2020)。3、HRM高分辨熔解曲线分析技术
高分辨率熔解曲线(High-resolution melting curve,HRM)由美国犹他大学分子病理学教授Carl Wittwer发明,是以PCR技术为基础,针对目标片段进行基因变异检测和分型的技术。HRM主要依赖于含有饱和DNA双链荧光染料、目的片段设计优化了的PCR靶标片段,能够有效控温获取高分辨熔解曲线信号的仪器系统和熔解曲线软件分析系统。该技术因其操作简单、检测成本低、速度快、PCR后无需开管,避免二次污染机会、检测灵敏度高等诸多优点已在医学和其他生物相关科学各类基因检测中迅速推广应用。
HRM主要原理为利用饱和型双链DNA(dsDNA)染料可插入PCR产物的特性,通过实时监测升温过程中dsDNA解链,荧光染料脱落,荧光信号减弱或消失的过程,高分辨记录熔解曲线,从而对单个碱基进行高分辨,对样品突变、SNP、甲基化位点进行高灵敏度分析。
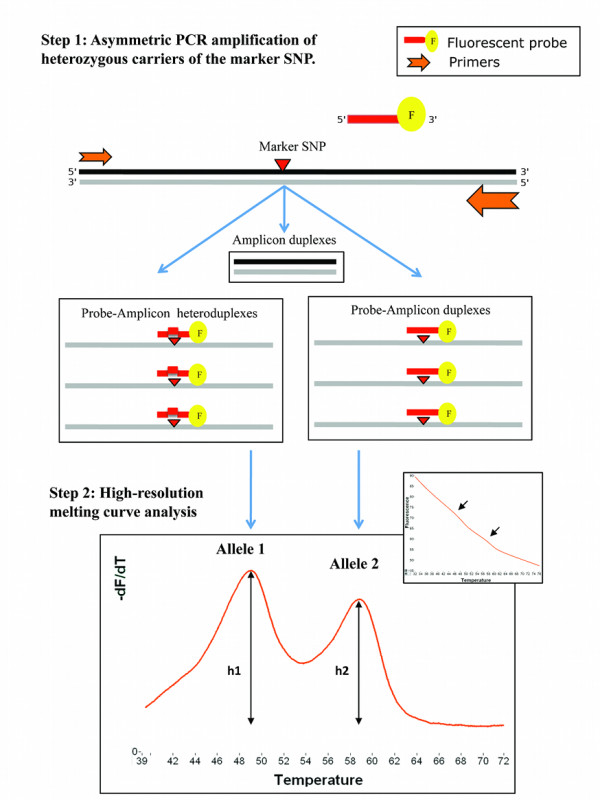
图4 用于检测等位基因表达不平衡的HRM原理示意图(Nguyen-Dumont et al., 2011)。
同其他检测基因突变的方法一样,HRM技术也存在一定的局限性,如难以辨别未知突变位点,检测灵敏度受检测基因片段SNVs类型影响。但该技术具有的优势恰好契合了现在个体靶向治疗分子诊断的需求。该技术不但同其他以目的基因片段为检测目标的基因检测方法一样,具有灵活、快速、灵敏度高、成本低等优点,还可实现全部基因变异类型的检测,而且可以灵活地增加检测通量,节约检测样品量、检测时间和成本、降低污染风险以满足临床检测需求。4、等温扩增技术
等温扩增技术(Isothermal Amplification Technology,IAT)也是基于PCR技术发展起来的一种在恒温下进行DNA扩增的分子生物学方法,与传统PCR相比,其不需热循环仪器,具有快速灵敏、简单便携、特异性强等优点,在分子诊断,尤其是床旁检测及实地筛查方面具有非常好的应用前景。其基本原理是利用异链替换酶、聚合酶和RNA剪切酶等多种酶基于循环式DNA扩增机制,在恒温条件下完成DNA扩增的过程。在等温扩增过程中,通过设计合适的引物,可以在RNA剪切酶的作用下产生长度不同的RNA片段。通过对这些RNA片段的检测,可以分析出DNA序列中的特定突变(Boonbanjong et al., 2022)。
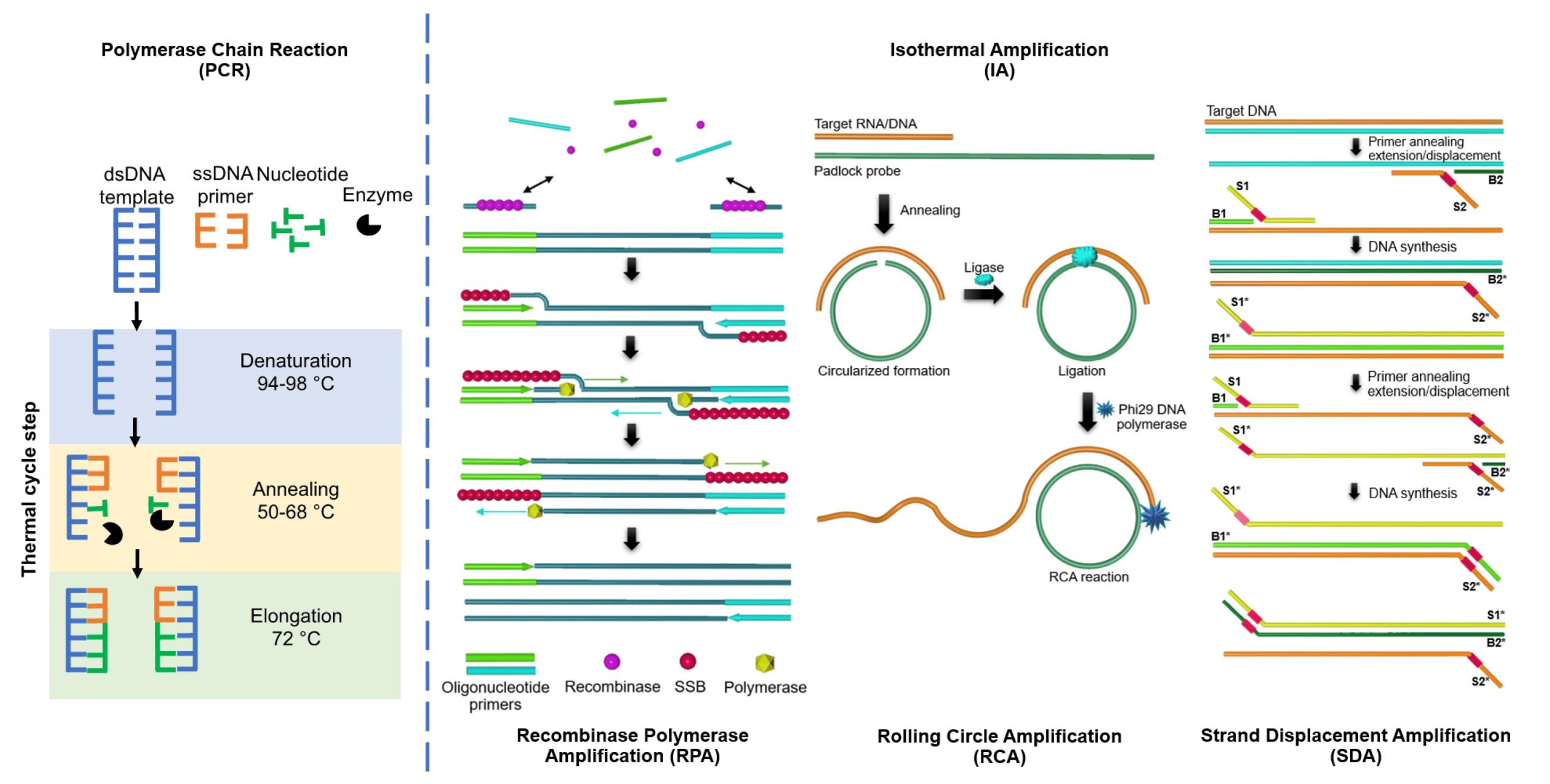
图5 PCR和一些IAT系统示意图(Boonbanjong et al., 2022)。
随着生物科学技术的发展,IAT已衍生出多种技术,如:环介导等温扩增(Loop-mediated isothermal amplification,LAMP)、滚环扩增技术(Rolling circle amplification,RCA)、依赖核酸序列扩增(Nucleic acid sequence-based amplification,NASBA)、依赖解旋酶DNA等温扩增技术(Helicase-dependent isothermal DNA amplification,HDA)、重组酶聚合酶扩增(Recombinase polymerase amplification,RPA)、切口内切酶恒温扩增反应(Nicking enzyme amplification reaction, NEAR)、链置换恒温扩增(Strand displacement amplification,SDA)、杂交链式反应(Hybridization chain reaction,HCR)等多种技术,由于篇幅有限,本文中不再详细讲解各类技术的原理及应用,对IAT感兴趣的同学可自行查阅相关资料进行学习。
更多关于PCR技术的讲解详见“PCR、qPCR、RT-PCR、RT-qPCR、Real-Time PCR你真的能区分的开吗?”一文。
三、基因测序法
基因测序的本质就是使用基因测序仪来确定基因上四种碱基(A、T、G、C)的排列顺序,其具体原理是用不同颜色的荧光标记基因中四种碱基,用基因测序仪里的激光系统按照碱基排列的顺序依次激发碱基发出荧光,碱基类型不同,荧光的颜色就不同,用专门的照相机捕捉荧光信号,确定基因上的碱基排列顺序。随着科学技术的进步,基因测序已从最初的一代测序,发展到后来的二代测序(NGS),以及现如今的三代测序(TGS)。1、一代测序
1977年,Sanger和Gilbert分别提出双脱氧链终止法和化学降解法,标志着第一代测序技术的诞生。其中Sanger双脱氧链终止法(即Sanger测序)是一代测序技术中最为经典的一种,也是目前应用最广泛的基因突变检测方法之一,而化学降解法如今已被逐渐遗忘。Sanger测序通过采用特异引物靶向模板DNA的特定区域,在PCR反应加入双脱氧核苷酸ddNTP,电泳分离延伸产物,对荧光标记进行检测,得到每个位置的碱基信息,进而分析出DNA序列中的特定突变(图6)。Sanger测序作为测序的“金标准”,其测序的准确率高于NGS和TGS,对700-1000bp读长具有较高的准确性,并且可以很好地处理重复序列。缺点是一次只能检测一个模板,速度慢且耗时(Zhang et al., 2021)。
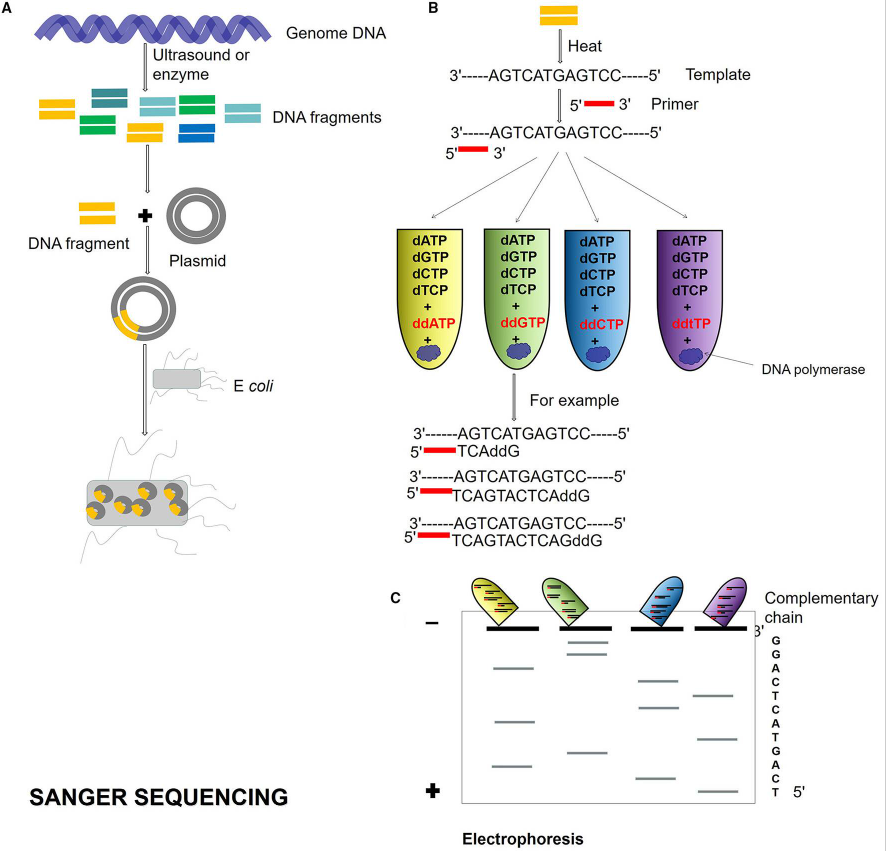
图6 Sanger测序过程(Zhang et al., 2021)。2、二代测序
二代测序(NGS)是建立在PCR技术和基因芯片基础上的,采用大规模并行测序技术,可同时合成数百万条互补链的测序模板并获取序列数据。NGS主要采用边合成边测序(SBS)技术和边连接测序(SBL)技术。目前,Illumina测序系统已成为NGS的主流产品。NGS在DNA复制过程中通过捕捉新添加碱基所携带的特殊标记(一般为荧光分子标记)来确定DNA的序列,其整体流程可以分为文库制备、生成DNA簇、边合成边测序和数据分析四个主要步骤。NGS有两个重要特点:高通量、读长短,因其高效和低廉的单碱基测序成本为临床应用提供了不可估量的前景优势。3、三代测序
三代测序(TGS)又称从头测序技术,是测序技术的一个里程碑,是基于单分子测序技术(Single Molecule Sequencing,SMS)和大规模并行测序技术发展起来的兴新技术,其测序时DNA分子无需PCR扩增,而是基于单分子的电信号或化学反应信号检测,实现了对每一条DNA分子的单独测序,TGS主要包括PacBio技术和Nanopore技术(Athanasopoulou et al., 2021)。以Nanopore系统为例,将纳米孔蛋白固定在电阻膜上,当核酸通过纳米孔时使电荷发生变化。由于纳米孔的直径非常细小,仅允许单个核酸聚合物通过,因此不同碱基通过蛋白纳米孔时对电流产生的干扰不同,通过实时监测并解码这些电流信号便确定碱基序列。TGS具有测序读长较长、可直接对原始DNA样本进行测序、可直接检测RNA及甲基化的DNA序列、运行快等优势。
四、杂交法
1、荧光原位杂交
1986年科研工作者开始利用异硫氰酸盐荧光素来标记探针,并在荧光显微镜下进行观察分析,建立了荧光原位杂交技术(Fluorescence in situ hybridization,FISH),将外源核酸(也就是常说的分子探针)与组织、细胞上待检测的DNA或RNA按照碱基互补配对原则进行配对,形成核酸杂交分子。将核酸探针的某一种核苷酸标记上报告分子(如生物素、地高辛),可利用该报告分子与荧光素标记的特异亲和素之间的化学反应,促使探针信号被荧光显微镜捕获,从而分析待测样本是否发生基因突变。2、基因芯片
基因芯片(Gene chip)技术是90年代中期以来得到快速发展的分子生物学高新技术,是各学科交叉综合的崭新科学,目前主要应用于疾病的诊断与治疗和药物研究两大方向。其主要原理为通过与一组已知序列的核酸探针杂交进行核酸序列测定的方法,在一块基片表面固定了序列已知的靶核苷酸的探针。当溶液中带有荧光标记的核酸序列TATGCAATCTAG,与基因芯片上对应位置的核酸探针产生互补匹配时,通过确定荧光强度最强的探针位置,获得一组序列完全互补的探针序列,据此可重组出靶核酸的序列。
参考文献
Athanasopoulou K, Boti M A, Adamopoulos P G, et al. Third-generation sequencing: the spearhead towards the radical transformation of modern genomics[J]. Life, 2021, 12(1): 30.
Boonbanjong P, Treerattrakoon K, Waiwinya W, et al. Isothermal amplification technology for disease diagnosis[J]. Biosensors, 2022, 12(9): 677.
Cao Y, Yu M, Dong G, et al. Digital PCR as an emerging tool for monitoring of microbial biodegradation[J]. Molecules, 2020, 25(3): 706.
Nguyen-Dumont T, Jordheim L P, Michelon J, et al. Detecting differential allelic expression using high-resolution melting curve analysis: application to the breast cancer susceptibility gene CHEK2[J]. BMC Medical Genomics, 2011, 4: 1-10.
Peng B, Wang Q, Luo Y, et al. A novel and quick PCR-based method to genotype mice with a leptin receptor mutation (db/db mice)[J]. Acta Pharmacologica Sinica, 2018, 39(1): 117-123.
Ye S, Dhillon S, Ke X, et al. An efficient procedure for genotyping single nucleotide polymorphisms[J]. Nucleic acids research, 2001, 29(17): e88-e88.
Zhang L, Chen F X, Zeng Z, et al. Advances in metagenomics and its application in environmental microorganisms[J]. Frontiers in microbiology, 2021, 12: 766364.
