细颗粒物(PM2.5)增加肺癌风险的方式,我们之前的理解可能错了。
近日,由英国弗朗西斯·克里克研究所Charles Swanton领衔的国际团队,在顶级期刊《自然》以封面论文的形式发表重磅研究成果,揭开了PM2.5驱动肺癌的机制[1]。
他们发现,PM2.5没有在肺细胞中诱发新的促癌突变,而是促进大量巨噬细胞进入肺组织,释放IL-1β创造炎性环境,让肺部原本存在的携带致癌突变(EGFR或KRAS)的正常肺细胞恶变,成为肺癌的种子。
他们还分析了英国、加拿大、中国和韩国32957名肺癌患者的数据,发现仅3年的高PM2.5暴露可能就足以使EGFR突变驱动的肺癌发生。这或许在一定程度上也解释了为什么中国肺癌患者EGFR突变携带率较高。

论文首页截图
空气污染对人类的健康有重大威胁。
世界卫生组织在2021年发布的报告显示,空气污染每年造成700万人死亡,其中99%的人生活在空气质量较差(年平均污染物超过5μg/m³)的地区[2]。
虽然已经有很多研究发现PM2.5与患心脏病或肺癌等疾病的风险增加发生有关[2],但是科学家对空气污染导致肺癌具体机制还缺乏清晰的认知。
要想解决问题,首先就得提出一个好的假设。对于空气污染导致肺癌的原因,Swanton团队有自己的想法。
他们注意到,最近三四年的一些研究数据表明,许多致癌物不会在肿瘤中留下可检测的DNA突变特征[4,5];而且在从不吸烟的肺癌患者(LCINS)肿瘤中,也没有发现外源性致癌基因突变[6,7]。
他们在分析TRACERx 421队列中的肺腺癌基因组后发现,在7-12%的吸烟者肺癌患者的肿瘤中也没有找到吸烟相关的致癌突变特征。
以上数据促使Swanton团队提出一个假设:空气污染物可能会促进肺组织微环境的炎症变化,使预先存在的突变克隆增殖,进而引发癌症。简单来说,空气污染没有诱发突变,而是把携带突变的健康肺细胞推上了癌变之路。

有了这个假设,研究方向一下子就变得清晰了。
他们选择研究由EGFR突变驱动的肺癌与空气污染之间的关系。原因很简单,EGFR突变在从不吸烟的肺癌患者中流行率很高,远高于吸烟的肺癌患者。因此,或许就是空气污染改变了肺组织的微环境,推动了EGFR突变驱动肺癌的发生。
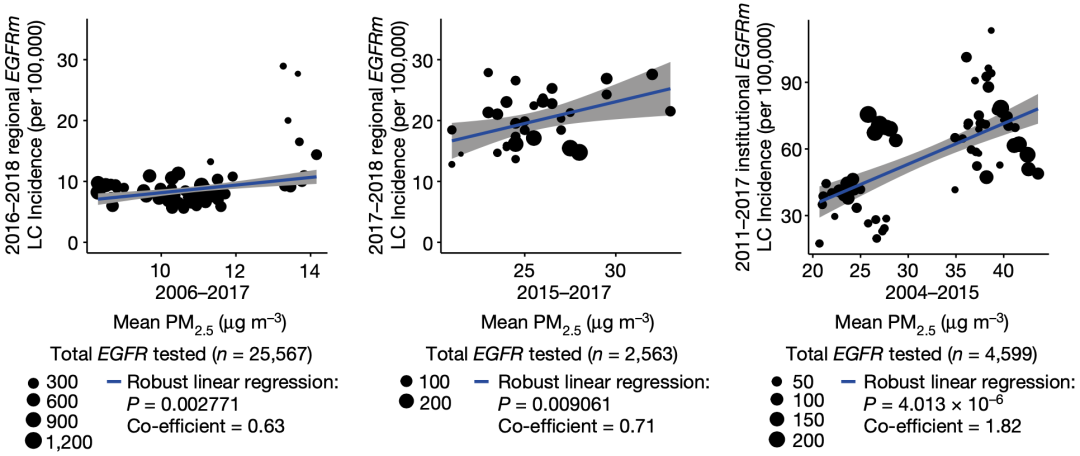
接下来,他们详细研究了英国、加拿大、韩国和中国台湾肺癌患者基因组数据和当地PM2.5数据,发现与低暴露(<6.77μg/m³)相比,高暴露(>7.27μg/m³)3年后,EGFR驱动的肺癌病例的频率明显更高(40% vs 73%,P=0.03)。
他们还分析了英国生物库中407509名参与者的数据,发现PM2.5水平与肺癌总体发病率有关(HR=1.08,P≤0.001)。这说明,空气污染对肺癌的驱动作用不限于EGFR驱动的肺癌。
基于以上研究结果,他们认为,EGFR驱动的肺癌发病率与PM2.5暴露水平之间存在关联,而且3年的空气污染暴露就足以让这种关联显现出来。
人体内发现的相关性,需要在小鼠身上揭示背后的机制。
Swanton团队让肺部表达EGFRL858R的肺腺癌小鼠模型,分别暴露于PBS、5μg和50μg的细颗粒物。他们发现与PBS对照组相比,露于5μg和50μg细颗粒物的小鼠肺部肿瘤负担更大,而且呈现出剂量依赖性。
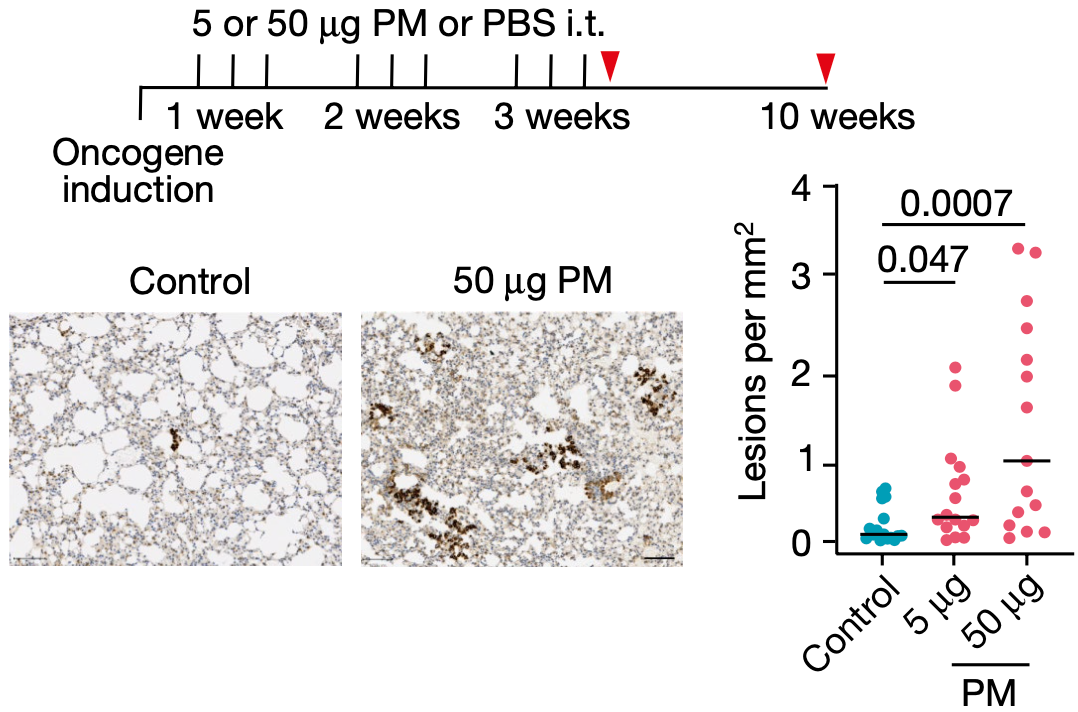
细颗粒物暴露导致小鼠肺部出现更多肿瘤
将EGFRL858R的表达限制在肺泡II型上皮细胞(AT2)内,细颗粒物也增加了肿瘤的负担。更让人吃惊的是,即使在EGFRL858R诱导表达之前暴露于细颗粒物也会增加肺部肿瘤负担。同样地,在KRASG12D肺癌小鼠模型中,暴露于细颗粒物也会增加肺部肿瘤负担。
这些数据共同表明,在致癌的KRAS和EGFR小鼠模型中,无论是在致癌基因表达之前还是表达之后暴露于细颗粒物,都可以促进肺部肿瘤的进展。
那暴露于细颗粒物之后,小鼠肺部的基因突变增加了吗?答案是没有。免疫系统在这个过程中又起到了什么作用呢?答案竟然是,必不可少。也就是说,细颗粒物促癌确实不是通过增加突变实现的,而是依赖于免疫系统,一旦小鼠免疫缺陷,细颗粒物就没有促癌作用了!
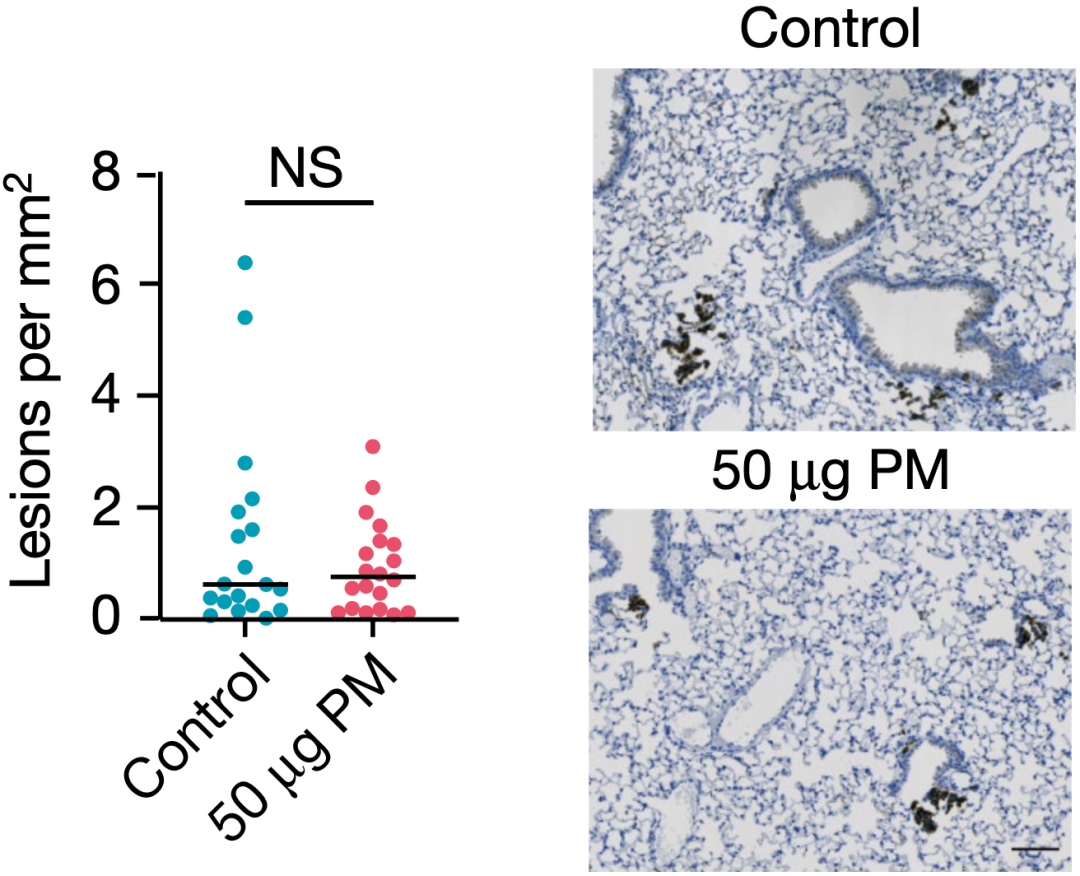
在免疫缺陷小鼠体内,细颗粒物不促癌
如此看来,肯定又是哪种免疫细胞帮了倒忙或者干脆叛变了。
介导有毒颗粒物诱发肺部局部免疫反应的巨噬细胞[9],进入Swanton团队的视线。而且研究数据也表明,短暂的细颗粒物暴露之后,浸润到肺部的巨噬细胞持续增加。几年前也有研究发现,肺部巨噬细胞在暴露于细颗粒物后,会释放炎症细胞因子[11]。因此,Swanton团队推测,巨噬细胞可能在细颗粒物促癌的过程中发挥着核心作用。
事实确实如此,Swanton团队发现,在细颗粒物的刺激下,巨噬细胞会释放IL-1β,IL-1β信号是促进细颗粒物介导EGFR驱动的肺癌所必需的,用抗IL-1β抗体阻断这一过程,可以抑制细颗粒物的促癌作用。
那么正常的肺上皮细胞暴露于细颗粒物之后会出现哪些变化呢?又为何会变成癌细胞呢?
基于肺组织的转录组数据,Swanton团队发现与AT2处于祖细胞(成体干细胞)状态和巨噬细胞招募有关的基因表达上调。他们回顾性分析一个人体研究的数据之后,也发现了类似的现象。也就是说,EGFRL858R AT2细胞在暴露于细颗粒物后,会发生转录重编程,进入有侵略性的祖细胞状态。值得一提的是,只有在EGFRL858R和细颗粒物同时存在的情况下,AT2才能进入祖细胞状态。
再结合前面巨噬细胞的研究数据,Swanton团队复原了细颗粒物诱发肺癌的全过程:在细颗粒物的刺激下,肺上皮细胞会招募巨噬细胞进入肺部,细颗粒物又刺激巨噬细胞释放IL-1β,导致EGFRL858R AT2重编程,进入祖细胞状态,成为启动肺癌的种子。
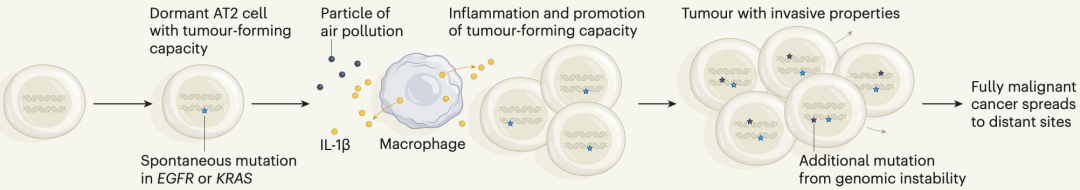
机制示意图
如此看来,携带EGFR或KRAS突变的人,如果生活在空气污染严重的地区,那就有些危险了。
为了初步了解人群中携带EGFR或KRAS突变的比例,Swanton团队分析了不同队列的监测数据。他们在一个有295例健康肺组织样本的队列中,发现54例(18%)携带有EGFR驱动突变;在81个健康肺组织样本中,发现43个(53%)携带有KRAS驱动突变。还有个更具体的数据是,在每554500个健康肺细胞中才有1个携带致癌性EGFR突变的细胞。此外,他们还发现,年龄与突变数之间存在显著的相关性。
总的来说,Swanton团队的这个研究再次证实了空气污染与肺癌之间存在因果关系,还破解了背后的机制,让我们对空气污染促肺癌有了更深入的了解。

《自然》封面
我还记得,大约在5年前,科学家开始关注健康组织中的体细胞突变情况。当时,他们的研究结果让我大吃一惊,因为研究数据表明几乎人人都携带基因突变,而且33%的人还携带有癌症相关突变[12,13]。
在一定程度上,Swanton团队再次证明了健康组织中确实存在致癌突变。更重要的是,Swanton团队的研究让我们意识到,这些因增殖而出现自发基因突变的正常细胞,在外界环境(例如PM2.5)的影响下,就有可能往恶性方向发展,导致癌症的发生。
如果随着年龄的增长,体细胞突变的发生和积累是不可改变的,那么我们确实该保护好我们的生存环境了,降低外界刺激增加癌症风险的可能性。另外,这个研究还有个预防癌症的启示,或许抗炎可以预防此类癌症的发生。
参考文献:
[1].Hill W, Lim EL, Weeden CE, et al. Lung adenocarcinoma promotion by air pollutants. Nature. 2023;616(7955):159-167. doi:10.1038/s41586-023-05874-3
[2].World Health Organization. WHO global air quality guidelines: particulate matter (PM2. 5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide: executive summary[J]. 2021.
[3].BERENBLUM I, SHUBIK P. A new, quantitative, approach to the study of the stages of chemical cartinogenesis in the mouse’s skin. Br J Cancer. 1947;1(4):383-391. doi:10.1038/bjc.1947.36
[4].Kucab JE, Zou X, Morganella S, et al. A Compendium of Mutational Signatures of Environmental Agents. Cell. 2019;177(4):821-836.e16. doi:10.1016/j.cell.2019.03.001
[5].Riva L, Pandiri AR, Li YR, et al. The mutational signature profile of known and suspected human carcinogens in mice. Nat Genet. 2020;52(11):1189-1197. doi:10.1038/s41588-020-0692-4
[6].Chen YJ, Roumeliotis TI, Chang YH, et al. Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell. 2020;182(1):226-244.e17. doi:10.1016/j.cell.2020.06.012
[7].Zhang T, Joubert P, Ansari-Pour N, et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat Genet. 2021;53(9):1348-1359. doi:10.1038/s41588-021-00920-0
[8].Myers R, Brauer M, Dummer T, et al. High-Ambient Air Pollution Exposure Among Never Smokers Versus Ever Smokers With Lung Cancer. J Thorac Oncol. 2021;16(11):1850-1858. doi:10.1016/j.jtho.2021.06.015
[9].Hogg JC, van Eeden S. Pulmonary and systemic response to atmospheric pollution. Respirology. 2009;14(3):336-346. doi:10.1111/j.1440-1843.2009.01497.x
[10].Ryu MH, Afshar T, Li H, et al. Impact of Exposure to Diesel Exhaust on Inflammation Markers and Proteases in Former Smokers with Chronic Obstructive Pulmonary Disease: A Randomized, Double-blinded, Crossover Study. Am J Respir Crit Care Med. 2022;205(9):1046-1052. doi:10.1164/rccm.202104-1079OC
[11].Hiraiwa K, van Eeden SF. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflamm. 2013;2013:619523. doi:10.1155/2013/619523
[12].Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362(6417):911-917. doi:10.1126/science.aau3879
[13].Yizhak K, Aguet F, Kim J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364(6444):eaaw0726. doi:10.1126/science.aaw0726
文章来源:奇点网
