1891年,Guido Werdnig 首次描述了一种致命的遗传性神经肌肉疾病:脊髓性肌萎缩症(SMA)。此后的一百多年来,众多杰出的科学家接力奋进,跨越认知迷雾,终于在最近10年里斩获了三款SMA疾病修正疗法。
在上个月举行的世界肌肉协会(WMS)2022年会上,三款药物中唯一的口服小分子药物利司扑兰,发布了JEWELFISH研究最新数据。研究显示,在6个月~60岁的经治SMA患者中,利司扑兰治疗24个月后,患者运动功能保持稳定,6分钟步行测试(6MWT)距离增加、疲劳感下降[1]。
JEWELFISH最新研究结果的发布,为成人SMA治疗领域带来了又一重要的证据。

这种异质性背后的原因很多,首先是基因的异质性。研究显示,95%的5q-SMA患者,SMN1基因存在7号/8号外显子纯合缺失[3],其余的患者存在SMN1基因的点突变,或者其他一些外显子上的小片段突变;同时患者携带SMN2基因的拷贝数也有差异。
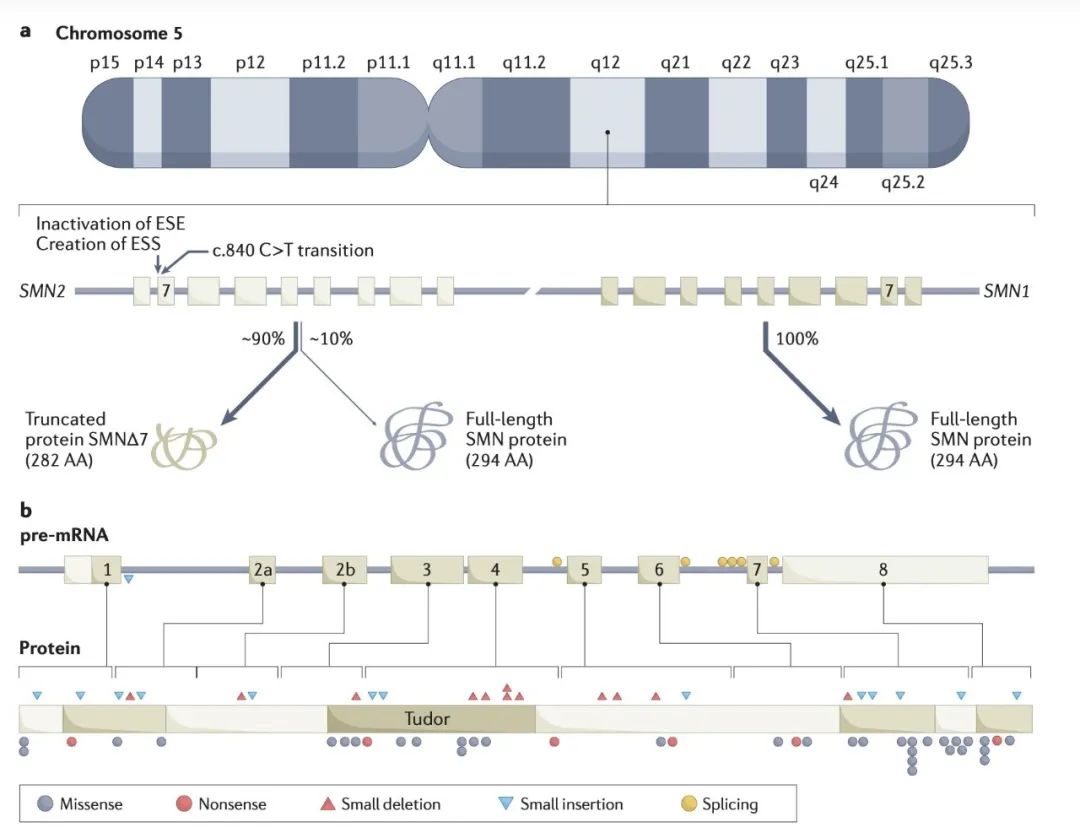
“SMN1和SMN2基因的转录也受各种因素的调控,包括转录因子,表观遗传学等等。近年来的研究还发现,其他一些基因,比如PLS3和NCALD,也是SMA的调控基因。” 福建医科大学附属第一医院神经内科陈万金教授告诉奇点网,“除此之外,病人的运动康复、体重管理、并发症管理、家庭护理,以及应对疾病的心态,都会对疾病的表型产生非常大的影响。”
面对这种异质性,JEWELFISH研究在纳入人群方面,尽可能贴近真实世界的患者情况。
JEWELFISH研究的患者人群涵盖1~3型,其中,1型占9%,2型占62%,3型占29%。SMN2基因的拷贝数也涵盖1~4个,其中1~2个拷贝占10%,3个拷贝占78%,4个拷贝占13%[1]。
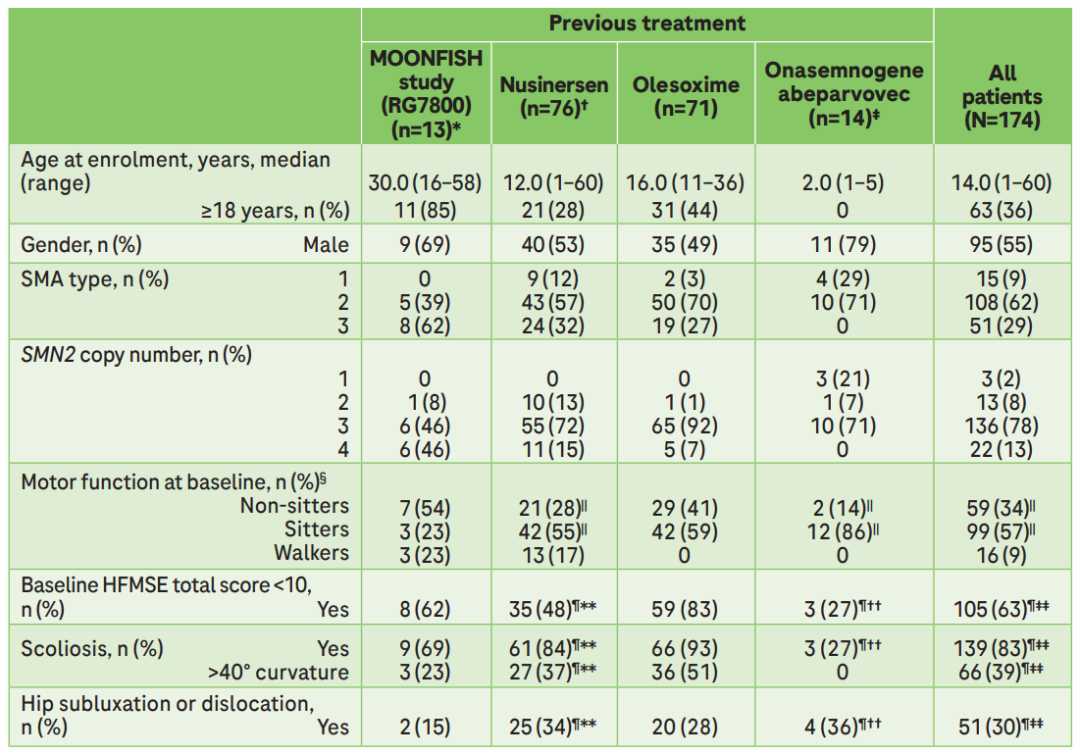
JEWELFISH研究人群广泛且异质性高,基线运动功能障碍程度高
“不同分型和表型的患者,对于治疗的预期也不同,但是不管分型如何,SMA治疗的准则都是一样:要早发现、早诊断、早治疗、早干预、早康复,这样患者的预后才会更好一些”,陈万金教授进一步解释,“很多遗传性的神经肌肉疾病,包括SMA在内,如果不进行治疗,患者的整体情况是走下坡路的。虽然有的成年患者在一定阶段,疾病可以保持稳定,但是碰到感染外伤等应激条件,可能出现疾病的急剧下滑。”
既往的研究显示,除了患有2型和3型SMA的儿童,可以在疾病早期阶段获得新的运动技能,并显示肌肉力量暂时增加之外,随着时间的推移,所有类型的SMA患者都会出现肌肉力量下降和运动技能进行性丧失[4]。从运动量表评分来看,神经肌肉医学研究委员会 (MRC) 总和评分每年平均损失至少1分,Hammersmith功能性运动量表扩展版 (HFMSE)损失至少0.5分。
“所以我认为像SMA这样的遗传性神经肌肉疾病,第一步就是要踩住刹车,阻止疾病的进展,不然疾病的列车就会一直往前冲”,陈万金教授补充道,“只有保住了患者即存的运动功能,才有可能争取更好的未来。这看上去好像是一个小目标,但其实意义非常重大。”
在JEWELFISH研究中我们看到,经治的SMA患者在接受利司扑兰治疗24个月后,运动功能保持稳定(评估量表包括:运动功能量表-32(MFM32)、上肢模块测试修订版(RULM )、HFMSE)。这表明,利司扑兰治疗可以扼制SMA自然病程中患者运动功能的衰退。
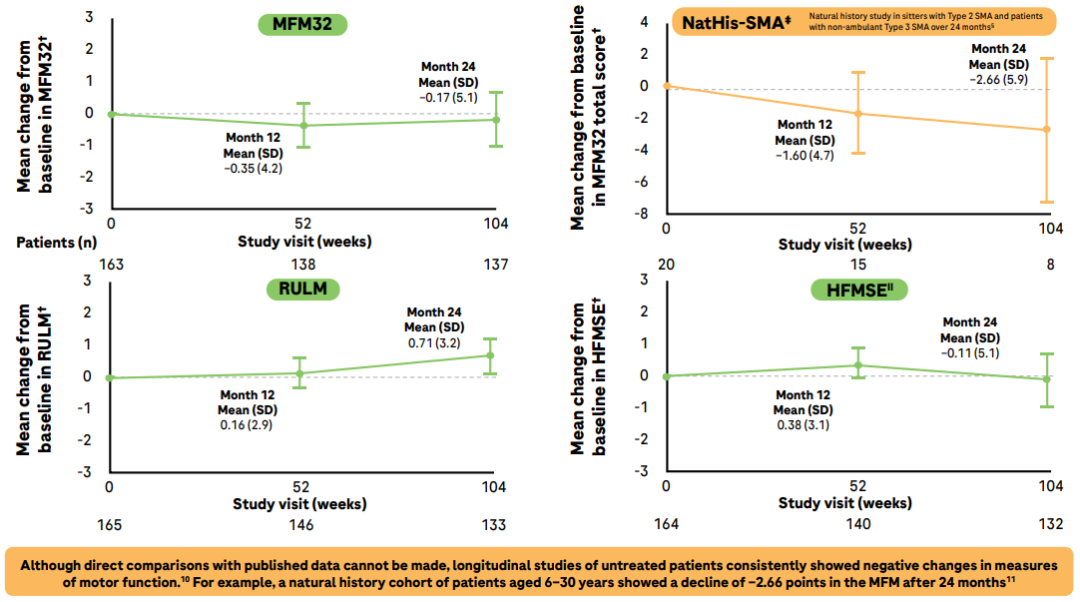
在2-60岁经治SMA患者中, 接受利司扑兰治疗24个月时运动功能稳定
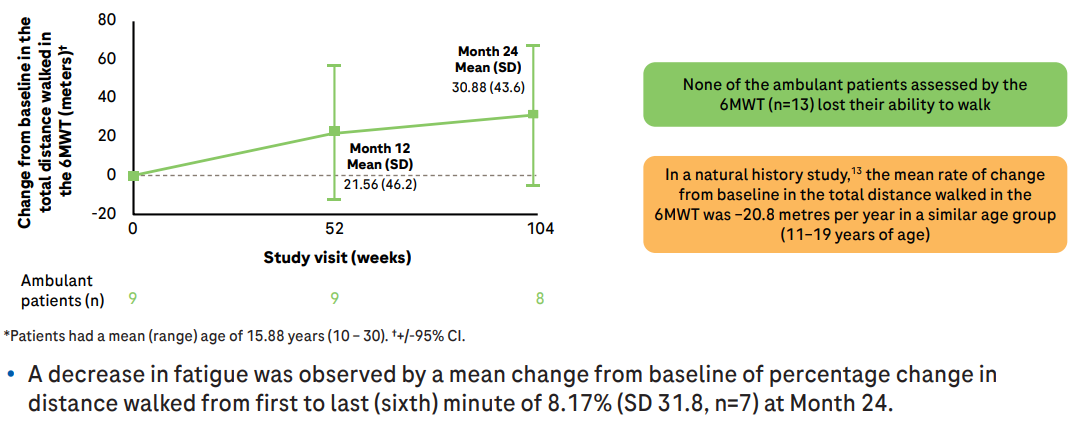
同时,可行走患者的6MWT距离增加,行走时的疲劳感也有所降低。要知道,在自然病程中, 2型和3型SMA患者6MWT每年分别减少20.8米(11~19岁)和9.7米(≥20岁)。
“在成人SMA治疗过程中,医生一定要和患者详细沟通治疗预期,同时有条件的可以继续一系列的家庭康复” ,陈万金教授告诉奇点网,“此外,成人治疗的给药方式也很重要,对于一些脊柱侧弯比较严重的病人,使用鞘注可能会存在一些困难,需要选择合适的药物进行治疗。”
在本次WMS大会上,由斯坦福医学院领衔的一项研究,评估了ATTEND量表的信效度。ATTEND量表用于不可坐立者和身体虚弱的能坐者,旨在评估微小但意义重大的运动功能改变。Rash信效分析发现,对于不可坐立者和身体虚弱的能坐者,ATTEND量表是一种可靠有效的评估方法,没有天花板或地板效应[5]。

此外,哈佛医学院领衔的一项研究,报告了SMA患者自我报告结局(SMA-PRO)的初步开发工作。研究定义了四个主要的事件领域,包括移动、轮椅、日常活动、步行辅助工具,通过对照护者或者患者本人进行访谈,更细致全面地评价SMA患者工具性生活能力表现[6]。

对于这两项研究,陈万金教授认为,“目前在成人SMA领域,其实每种量表都有它的局限性。上面提及的两个研究,一个是用相对客观的数据去评估患者的微小运动改善,另一个是收集患者和照护者的主观反馈,都是非常好的尝试,可以进一步完善成人SMA治疗的评估方式。”
近年来,随着SMA疾病修正疗法的进一步覆盖,对于成人SMA患者,亟需更完善的量表去评价有意义的运动功能改善。尤其值得注意的是,越来越多的专家倡议,将患者和照护者的体验纳入评估体系,因为一些微小的局部改变,可能在临床上不具备重大意义,但可能为患者的生活带来巨大的改变。
“比如患者一根手指的力度增加了,带来的变化也可能是惊人的,他可以自己驱动电子轮椅,可以打字,使用手机和电脑,这相当于给他打开了一个通往虚拟世界的大门,”陈万金教授进一步解释,“我们知道,SMA患者都很聪明,一旦他们可以通过网络和世界沟通,就可以打开自己思维的疆域,在虚拟世界做很多事情,包括创造财富。”
自2016年第一款SMA疾病修正疗法上市以来,目前已经有三款药物获得了美国FDA批准。在最上游基因层面,有针对SMN1的基因治疗,针对SMN2的前体mRNA剪接调节,以及正在研究中的基因编辑。在中游的蛋白层面也有不少研究,例如靶向肌肉生长抑制素的在研药物;而在下游,也有一系列涉及神经元抗凋亡和再生的研究。
“当然还有很多跨学科的进展,比如神经重塑技术,脑机接口技术等等,未来这些技术都有可能进一步改善SMA患者的处境。我们的研究团队也正在进行基因编辑治疗相关研究,和世界范围内的科学家一起努力攻克SMA。所以对于SMA患者来说,当务之急是控制疾病进展,保持面对未来的信心。”
陈万金教授还向奇点网透露,目前他们的研究团队可以为全国范围内,包括SMA在内的神经遗传病患者提供免费的基因检测服务,帮助进一步提升这些疾病的诊断。
“相信在大家的共同努力下,SMA患者的生存境遇会越来越好!”
参考文献:
[1]JEWELFISH: 24-month safety, pharmacodynamic and exploratory efficacy data in non-treatment-naïve patients with SMA receiving treatment with risdiplam . WMS 2022.
[2]Sansone, V. A., Walter, M. C., Attarian, S., Delstanche, S., Mercuri, E., Lochmüller, H., … Hagenacker, T. (2020, September 18). Measuring Outcomes in Adults with Spinal Muscular Atrophy – Challenges and Future Directions – Meeting Report. Journal of Neuromuscular Diseases. IOS Press.
[3]Mercuri, E., Sumner, C. J., Muntoni, F., Darras, B. T., & Finkel, R. S. (2022, August 4). Spinal muscular atrophy. Nature Reviews Disease Primers. Springer Science and Business Media LLC.
[4]Wadman, R. I., Wijngaarde, C. A., Stam, M., Bartels, B., Otto, L. A. M., Lemmink, H. H., … Pol, W. L. (2018, February 2). Muscle strength and motor function throughout life in a cross‐sectional cohort of 180 patients with spinal muscular atrophy types 1c–4. European Journal of Neurology.
[5]Adaptive Test for Neuromuscular Disorders: Design of a Wheelchair-Based Assessment .WMS 2022.
[6]Updates on the Development of the Spinal Muscular Atrophy – Person-Reported Outcome (SMA-PRO): A Caregiver and Self-Proxy Performance Measure for Children and Adults with SMA .WMS 2022.
文章来源:奇点网
