高温高湿的气候因素和高强度体力活动是导致热射病最主要的危险因素(刘树元等. 2019)。其中,经典型热射病主要由高温和(或)高湿环境因素引起,通常没有剧烈的体力活动;劳力型热射病主要由于高强度体力活动引起机体产热与散热失衡而发病。

图1 中暑症状与预防示意图。
具体而言,ZBP1是一种Z-核酸受体,其通过诱导RIPK3依赖性细胞死亡来介导中暑,而HSF1可增加ZBP1的表达,并通过不依赖于核酸传感作用的机制激活ZBP1,重要的是,ZBP1、RIPK3或MLKL和caspase-8的缺失降低了热应激诱导的循环衰竭、器官损伤以及致死率。团队的发现为热射病的治疗提供了全新思路。
那么团队是如何发现这一全新致病机制的呢?下面就和伯小医一起去走进团队的研究中,并学习一下这篇文章的研究思路和实验方法吧!
在此前的研究中,研究者发现,在脊椎动物中,程序性坏死是由MLKL或gasdermin家族蛋白介导的(Wallach et al., 2016)。其中,MLKL被RIPK3依赖性磷酸化激活并执行坏死性凋亡(图2);gasdermin D(GSDMD)的激活会引发细胞焦亡(图3)。
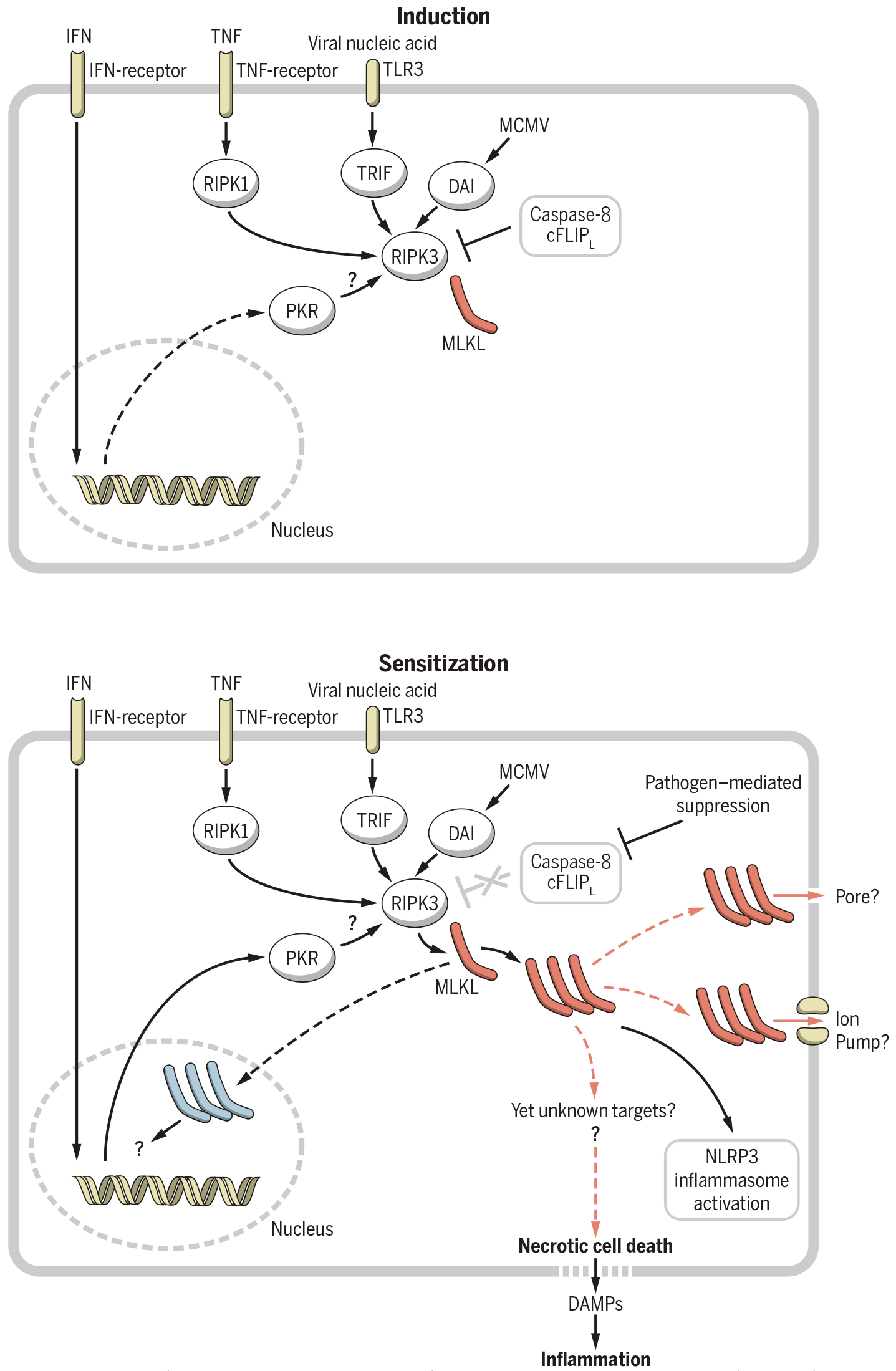
图2 启动坏死性凋亡信号传导的蛋白激活在两个机制水平上受到控制。第一个是诱导,主要通过细胞外宿主衍生的配体,如TNF介导(上图);第二个是致敏,由病原体和其他抗细胞抑制活性的试剂引起,允许MLKL磷酸化和寡聚化,从而引发坏死性细胞死亡(下图)。此外,该图还描绘了MLKL激活的各种分子后果,可能有助于诱导细胞死亡的标记为橙色(Wallach et al., 2016)。
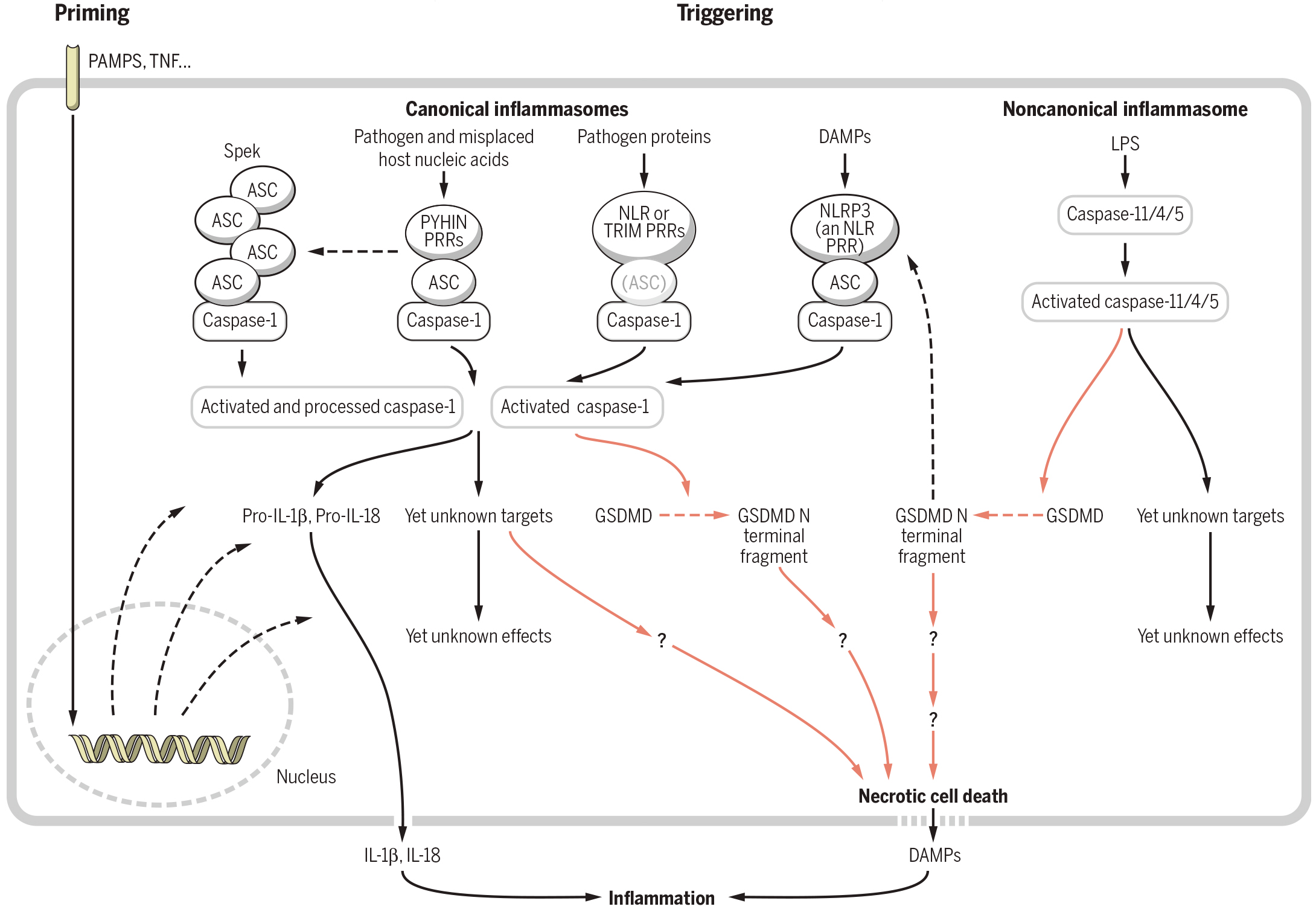
图3 细胞焦亡的信号传导在两个机制水平上受到调节,就像在坏死性凋亡的情况下一样,但以一种相反的方式(Wallach et al., 2016)。
RIPK3调控热应激诱导的细胞死亡和热射病症状
将Ripk3-/-、Mlkl-/-、Mlkl-/-Casp8-/-及相应野生型(WT)小鼠置于39°C、相对湿度60±5%的环境中以构建小鼠中暑模型。
热应激可使动物核心温度升高到43℃,并引起严重的电解质紊乱;诱导RIPK3和MLKL在肝、肺及肠中的磷酸化,且血清中RIPK3浓度、血清乳酸和促炎细胞因子浓度均升高;同时,血管内凝血酶生成、血小板聚集、纤维蛋白沉积、微循环阻塞和循环DIC标志物亦增加;并且,诱导pro-casp8、pro-casp3、GSDME和GSDMD裂解;而所有这些都可通过RIPK3基因缺失来缓解(图4A-G)。
有趣的是,RIPK3激酶活性的丧失不仅可消除热应激诱导的MLKL磷酸化,但不影响pro-casp8、pro-casp3或GSDME的切割以及热应激后死亡标志物的水平,还可拯救70%的小鼠热应激后的致死;同时,MLKL缺失减轻了器官损伤并拯救了75%的小鼠,而MLKL和casp8双缺可使所有小鼠都能在热应激中存活(图4H-I)。
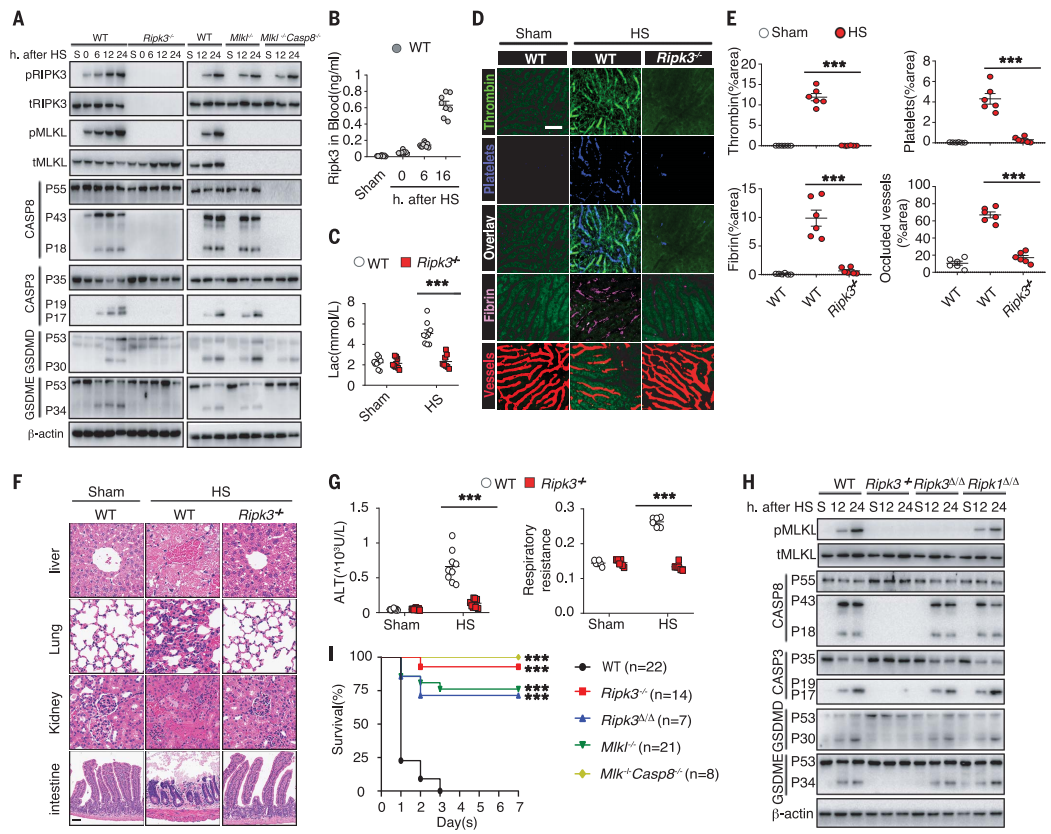
图4 RIPK3介导热应激诱导的细胞死亡和热射病的病理特征(Yuan et al., 2022)。
热应激通过激活RIPK3触发细胞死亡
将L929小鼠成纤维细胞暴露于43°C 2h或 42°C 6h。并提取WT或Ripk3-/-小鼠骨髓来源的巨噬细胞(BMDM)和腹膜巨噬细胞(PM),进行相同实验。
RIPK3敲除阻断了热应激诱导的RIPK3、MLKL磷酸化及pro-casp8、pro-casp3和GSDME水平(图5A),这与乳酸脱氢酶(LDH)实验检测的细胞死亡减少、细胞膜完整性丧失和细胞膜通透性增加有关(图5B、C)。而恢复RIPK3表达后,Ripk3缺陷型L929细胞响应热应激导致细胞死亡的能力得以恢复(图5D)。
在Mlk1-/-而不是Ripk3-/-BMDMs中,热应激暴露后24h内延迟了细胞死亡(图5E)。与体内实验相同,RIPK3激酶活性的丧失阻止了MLKL的磷酸化,但没有阻止casp8、casp3和GSDME的裂解,MLKL和casp8的缺失几乎完全阻断了casp3和GSDME的切割(图5F)。
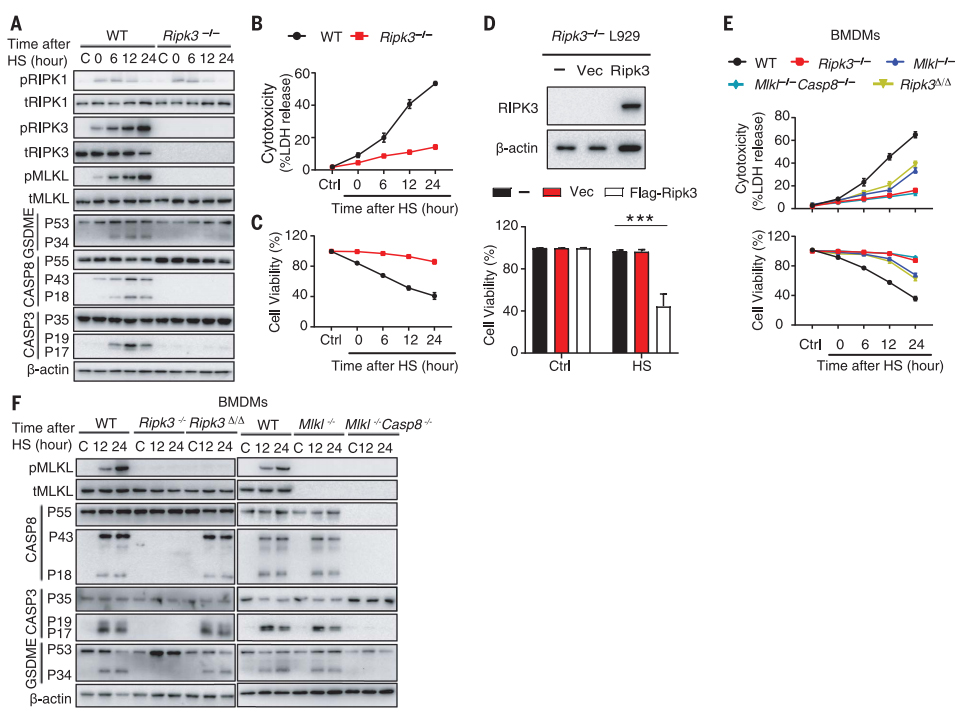
图5 热应激触发RIPK3依赖性细胞死亡。
热应激通过ZBP1激活RIPK3
探究RIPK3相互作用蛋白是否是热应激诱导细胞死亡所必需的。
TRIF缺失、RIPK1激酶结构域突变(RIPK1△/△)或通过necrostain-1抑制RIPK表达后均不影响热应激诱导的BMDMs细胞死亡,相比之下,ZBP1的缺失消除了RIPK3、MLKL的磷酸化及casp8、casp3和GSDME的裂解与细胞死亡(图6A、B);而ZBP1表达恢复后,ZBP1缺陷导致的热应激后细胞死亡能力得以恢复(图6C),该结果在HT-29细胞中得到了验证(图6D);并且,热应激诱导了ZBP1和RIPK3之间的相互作用(图6E、F)。
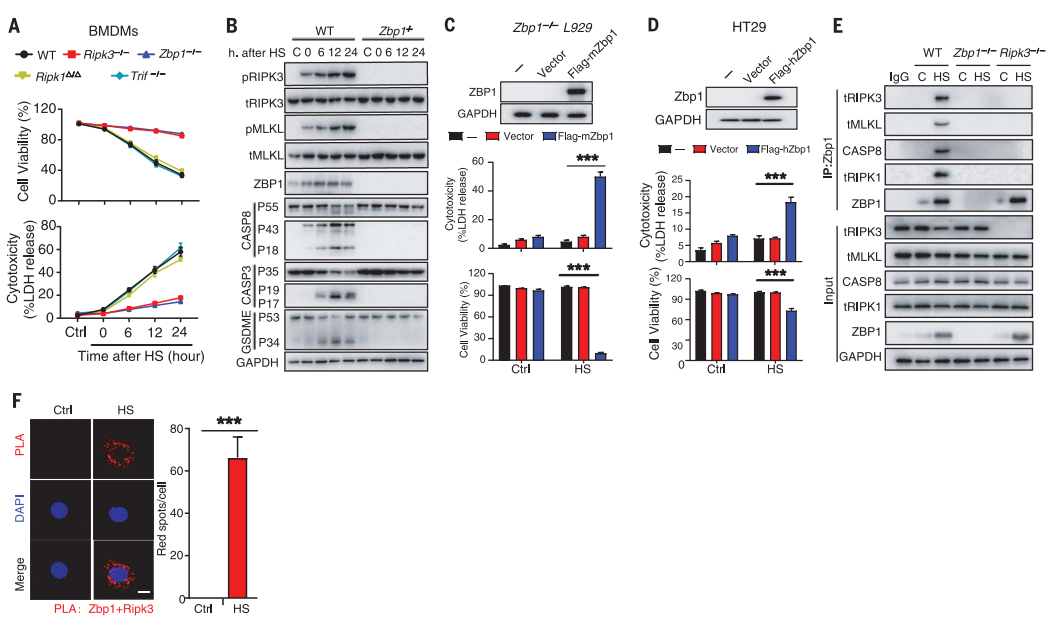
图6 热应激通过ZBP1激活RIPK3。
ZBP1介导热射病的病理特征
将Zbp1-/-小鼠及其WT同窝小鼠暴露于热应激以确定ZBP1是否激活RIPK3并在热应激后介导热射病。
ZBP1的缺失阻断了热应激诱导的RIPK3和MLKL的磷酸化以及CASP8、CASP3、GSDMD和GSDME在肝脏和肠道中的裂解(图7A);ZBP1的缺失可防止热应激诱导的DIC、全身炎症反应、循环衰竭、多器官损伤和致死性,这与RIPK3缺乏症相似(图7B-F);相比之下,TRIF的缺失或RIPK1激酶结构域的突变未能保护小鼠免受致命的热应激(图7G)。
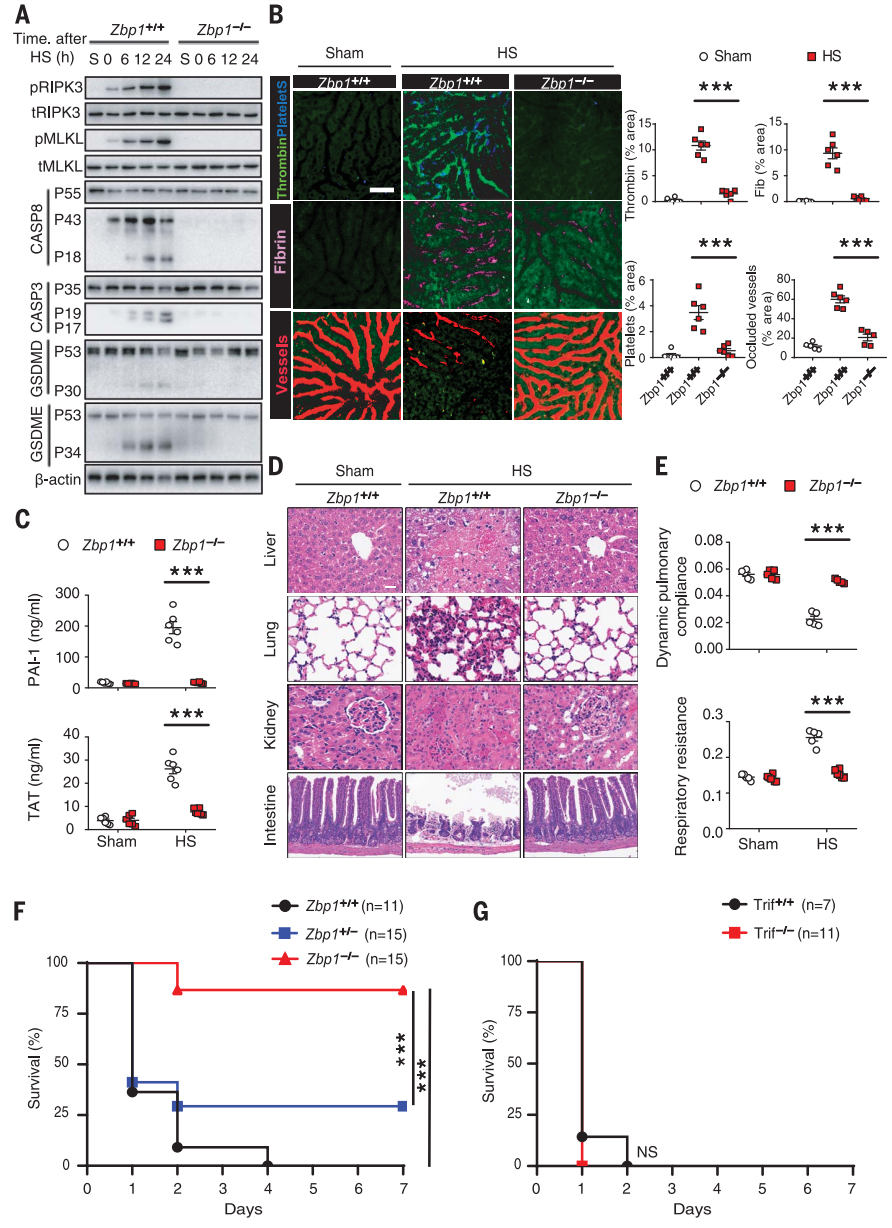
图7 ZBP1介导热应激诱导的细胞死亡和热射病的病理特征。
热激应通过调控HSF1增加ZBP1的表达
指定时间内受到热应激后,qRT–PCR与Western-blot分析体内外模型中ZBP1的表达水平;生信分析结合双荧光素酶报告与ChIP实验验证ZBP1启动子与HSF1的结合。
热应激可上调L929细胞和小鼠巨噬细胞中ZBP1的表达,其上调方式与1型干扰素类似,并刺激肺、肝、肾和肠中的ZBP1转录(图8A-E);确定HSF1的预测结合位点,并进行验证,热胁迫增强了ZBP1启动子中HSF1结合位点的激活和占用(图8F-G)。进一步实验表明,HSF1的缺失抑制了热应激诱导的ZBP1表达和细胞死亡的增加(图8H)。
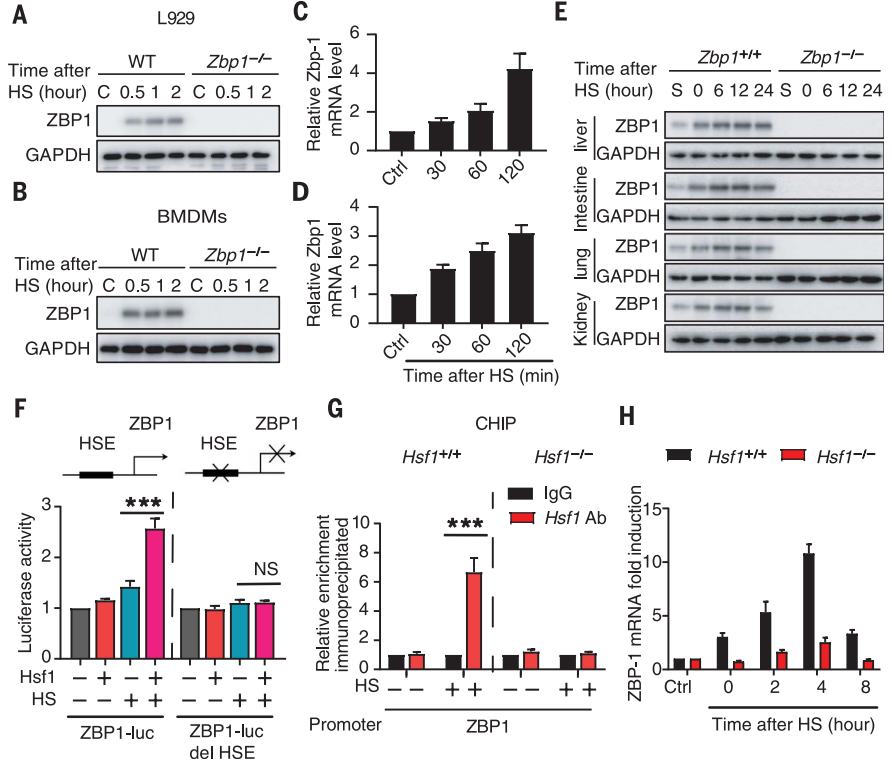
图8 热应激通过HSF1增加ZBP1的表达。
热应激通过RHIM结构域激活ZBP1,与Z-核酸感应无关
研究热应激促进ZBP1活化的机制,构建表达完整的ZBP1或ZBP1突变体的转基因L929细胞株,这些突变体要么缺少Zα,Zα1或Zα2结构域,要么包含Zα2结构域内的点突变(图9A)。
热应激仍会在表达ZBP1突变体的L929细胞中诱发ZBP1-RIPK3相互作用、RIPK3和MLKL磷酸化及细胞死亡(图9B-E)。
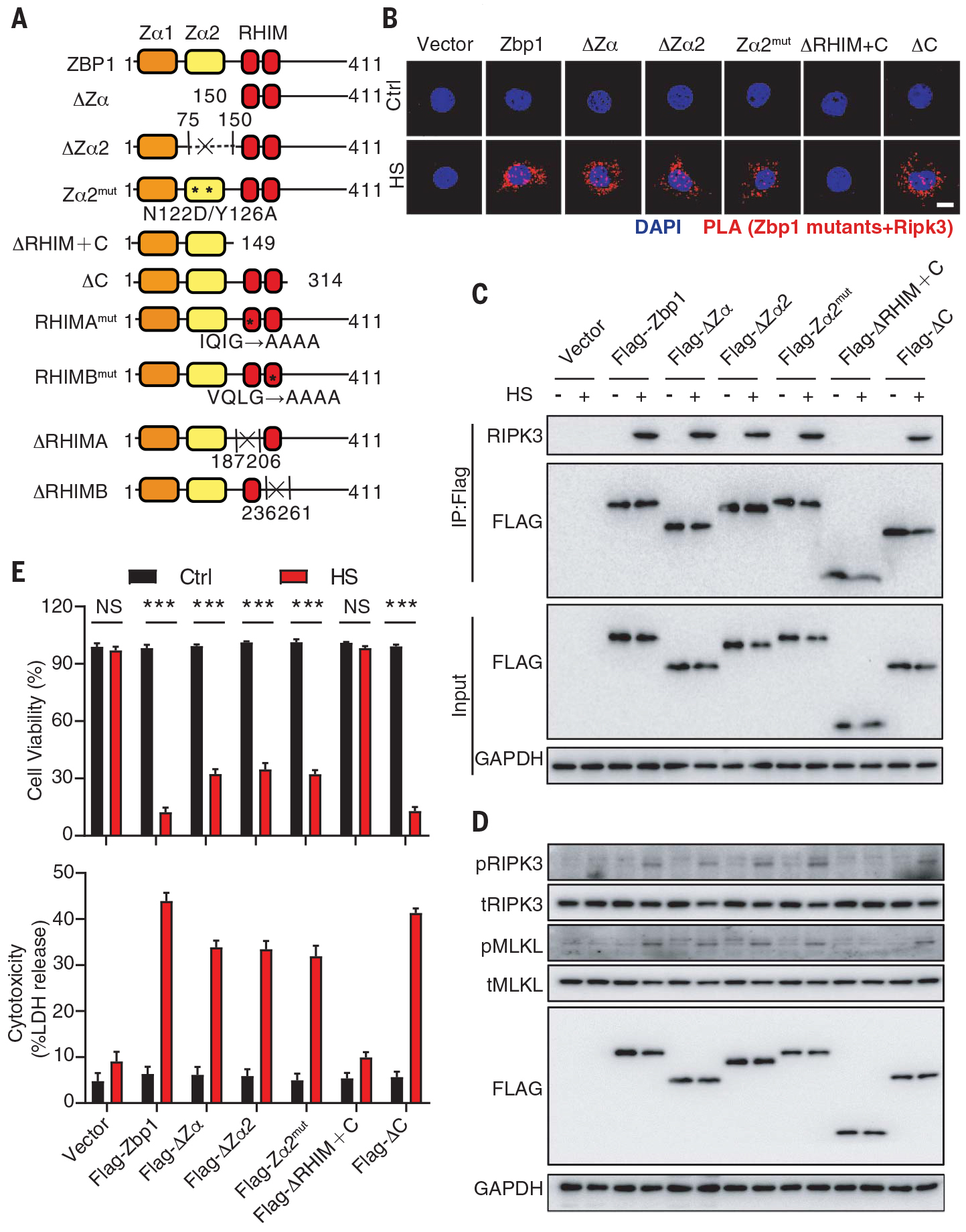
图9 热应激通过RHIM结构域激活ZBP1,与Z-核酸感应无关。
最终结论
本研究确定了ZBP1可通过RIPK3依赖性细胞死亡促进热射病病理特征的作用。
虽然,热应激会增加ZBP1的表达,但其作用机制与Z-核酸感应无关,也就是说,ZBP1在热胁迫下的激活是可有可无的。
ZBP1的RHIM-A结构域依赖性聚集又可能利于热应激诱导的细胞死亡,内源性Z-核酸可能会增强热应激诱导的ZBP1活化。持续的环境热暴露导致的极端高温可能会致使ZBP1依赖性细胞死亡过度激活,最终诱发器官发生循环衰竭、DIC、多器官功能障碍,甚至死亡。
Wallach D, Kang TB, Dillon CP, et al. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352(6281):aaf2154.
Yuan F, Cai J, Wu J, et al. Z-DNA binding protein 1 promotes heatstroke-induced cell death. Science. 2022;376(6593):609-615.
刘树元,宋景春,毛汉丁,等. 中国热射病诊断与治疗专家共识[J]. 解放军医学杂志,2019,44(03):181-196.
