之前坏死性凋亡专题里的一篇文章,我们讲解了一篇关于抗肿瘤微环境中,坏死性凋亡的关键蛋白RIPK3通过磷酸化TRIM28解除其抑制作用,从而激活NF-κB转录活性,增强免疫调节因子表达的文章,相信大家对坏死性凋亡的抑癌机制已经有了初步认识。这次我们从坏死性凋亡的另一条诱导途径入手,来聊一聊怎样将肿瘤治疗与坏死性凋亡表型结合,从而将基础研究进行转化,解决我们临床中遇到的问题。
一般我们所熟悉的坏死性凋亡通路是TNF诱导的RIPK1依赖通路。然而除了以RIPK1激活为标志的坏死性凋亡外,还有一些诱导因素以RIPK1非依赖的模式启动坏死性凋亡,比如双链核酸(dsDNA和dsRNA)。这些双链核酸通常来源于病毒或内源性逆转录病毒元件(EREs),活化感受器蛋白ZBP1,启动下游RIPK3的激酶功能和坏死小体形成。
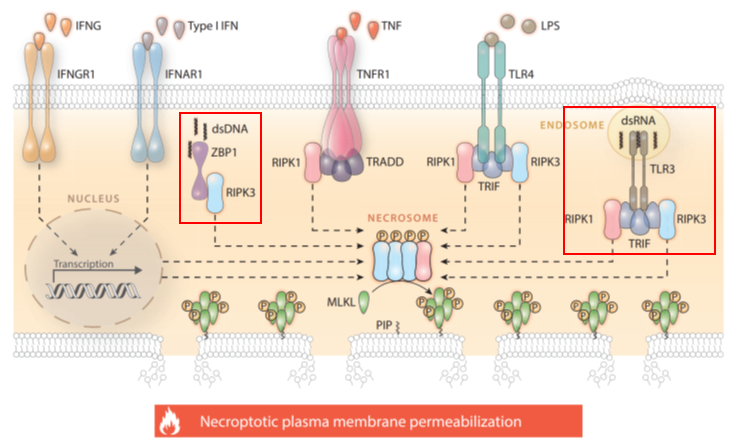
(图片来源:DOI:10.1146/annurev-pathol-052016-100247)
这篇近期发表于Nature的论文就是聚焦于一种特殊形态的双链RNA–Z型RNA(左手螺旋),揭示RNA编辑酶ADAR1阻断Z-RNA形成,以及随之而来的ZBP1依赖性坏死性凋亡介导的免疫反应机制,通过这一原理筛选能够促进Z-DNA(同样激活ZBP1)积累的小分子化合物,诱导肿瘤浸润成纤维细胞ZBP1依赖的坏死性凋亡,最终将黑色素瘤的“冷”肿瘤(免疫浸润低)转换为“热”肿瘤(免疫浸润高),解决免疫检查点治疗抵抗的问题。

本文是从机制研究到临床转化的典范,适合想要进行机制研究又想结合临床转化的研究者参考。
全文主要分为两大块内容,第一大块是机制研究部分,第二大块是小分子筛选和体内外功能验证的部分,前者是后者的基础,后者是前者的延展。由于篇幅所限,这里不为大家讲解每个结果的细节内容,但求帮助大家理解思路。
一、机制研究
本文机制研究的部分主要解决的问题是:ADAR1是否抑制细胞内源性Z-RNA的积累,抑制Z-RNA激活ZBP1介导的细胞程序性死亡,以及具体通过何种方式产生这一效应。因此机制研究主要为体外细胞研究,分为两部分:
1.构建ADAR1敲除和位点突变细胞,检测Z-RNA的变化和具体来源
首先需要探究的是ADAR1是否抑制Z-RNA生成,本文使用的是CRISPR/Cas9敲除的方法,因此问题就转换为敲除ADAR1后Z-RNA是否积累。通过免疫荧光染色可以看到,ADAR1敲除后使用IFNβ诱导,Z-RNA(左手螺旋)和A-RNA(右手螺旋)都在细胞中积累;若将ADAR1与Z-RNA的关键结合位点突变,则获得相同结果;但仅突变RNA编辑结构则效果不明显。进一步用RIP-seq测序手段检测Z-RNA的来源以及ADAR1结合的位置,发现ADAR1主要结合在ISG(IFN激活基因)mRNA的3’UTR区域。通过对结合RNA结构的解析,发现ADAR1结合的Z-RNA主要为包含SINE(短散在核元件)序列和GU重复序列的mRNA。一旦ADAR1对这些RNA进行编辑,则无法形成Z-RNA构象。
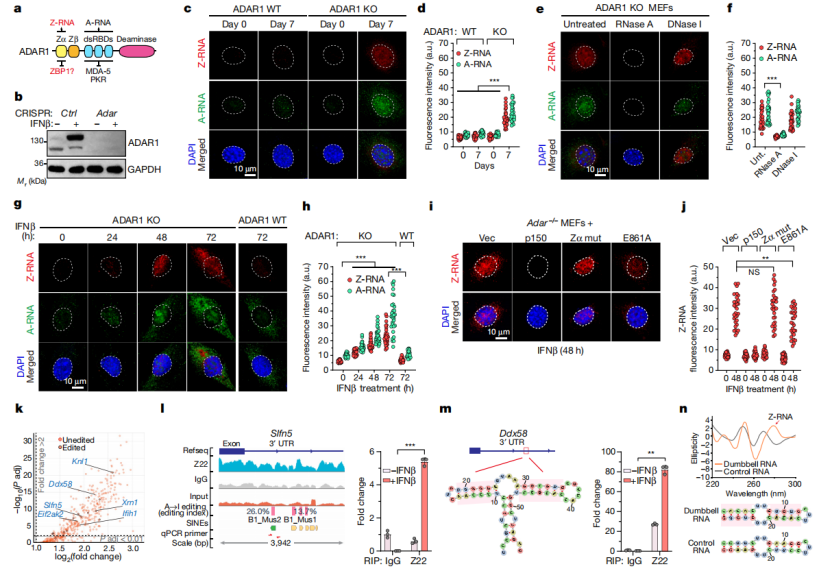
2.同时敲除ADAR1和ZBP1,检测ZBP1和Z-RNA的结合以及诱导的坏死性凋亡
这一部分比较简单和常规,主要是为了方便之后对ZBP1的研究,在细胞内敲除ZBP1,并敲除ADAR1或转染FLAG-ZBP1,通过邻位连接技术(PLA)、ChIP-seq和对坏死性凋亡标志物的检测,证明敲除ADAR1后,Z-RNA与ZBP1结合,诱导ZBP1依赖的坏死性凋亡(和细胞凋亡)产生。
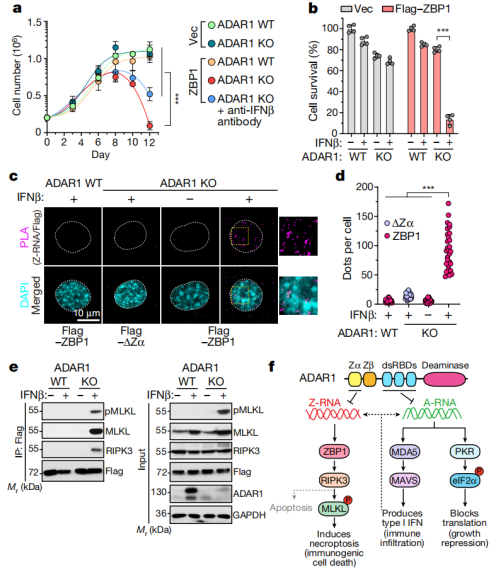
二、小分子药物筛选
基于前一部分得到“ADAR1抑制Z-RNA,敲除ADAR1能够提高Z-RNA积累,激活ZBP1诱导细胞程序性死亡”的结论,作者试图找到既能够避开ADAR1抑制作用,又能激活ZBP1的小分子化合物。因此想到了ZBP1不仅能够结合Z-RNA,也能结合构象类似的Z-DNA,而ADAR1并不能对DNA进行编辑,从而获得一箭双雕的效果。于是这一部分的临床转化研究就分为三部分:
1.促进Z-DNA积累的化合物筛选和验证(CBL0137)
这一部分是利用已知能够插入DNA,影响DNA拓扑结构或改变染色质结构和动力学的小分子化合物进行筛选,初步得到显著诱导Z-DNA积累的化合物CBL0137。与机制部分研究相似,通过Z-DNA抗体的ChIP-seq实验,发现CBL0137和DNA结合的位置,以及CBL0137浓度与Z-DNA比例的关系。
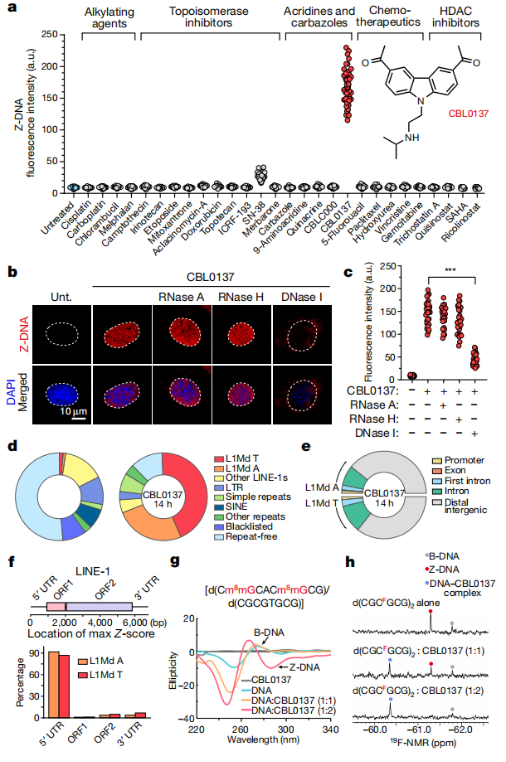
2.CBL0137体外处理验证Z-DNA诱导的ZBP1依赖坏死性凋亡
这一部分的实验也是我们最熟悉的体外功能验证部分。同样利用前文中提到的ZBP1敲除和ZBP1敲除+转染FLAG-ZBP1细胞,加入CBL0137的处理,检测细胞活性和坏死性凋亡指标;再通过ZBP1的ChIP-seq,验证其结合的Z-DNA与CBL0137诱导的Z-DNA有较大重合。最后进行坏死性凋亡p-MLKL的定位,表明CBL0137诱导的ZBP1坏死性凋亡为核裂解型(区别于RIPK1依赖的坏死性凋亡)。
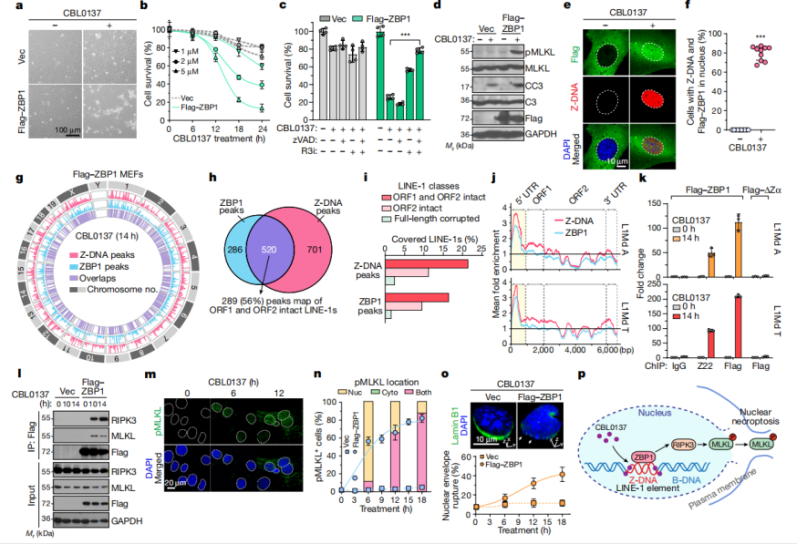
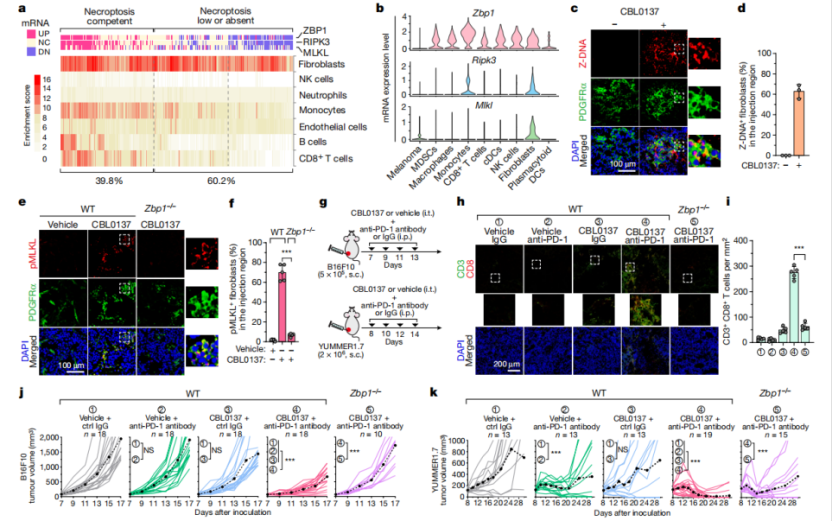
3.体内动物模型验证CBL0137诱导肿瘤浸润成纤维细胞坏死性凋亡,提高肿瘤免疫浸润,降低黑色素瘤ICB(免疫检查点抑制剂)抵抗
最终筛选到的CBL0137还需在体内验证功能。作者首先对公共TCGA数据库黑色素瘤数据进行免疫浸润分析和坏死性凋亡标志物表达量分析,再用GEO数据库中ICB抵抗肿瘤单细胞测序数据进行分析,发现肿瘤浸润成纤维细胞比例较高,且表达坏死性凋亡标志物。通过CBL0137组合PD-1抗体治疗,黑色素瘤生长显著受到抑制,坏死性凋亡标志物与成纤维细胞标志物共定位,T细胞浸润增加,而ZBP1敲除小鼠肿瘤无此现象。至此,完整的体内实验表明CBL0137可通过激活ZBP1依赖的肿瘤成纤维细胞坏死性凋亡,促进免疫浸润,而正常细胞由于低表达ZBP1而不受影响。
有读者会说,由于经费和平台因素,Nature的工作并不是广大普通课题组可以模仿的,那么我们该从这样的文章中学习些什么呢?
诚然,本文工作量大,涉及众多测序数据,但仍有部分内容我们可以参考。这里就为大家分析一下本文中我们可以借鉴的地方:
1.在思考研究方向时,可联系临床问题和科学问题(表型),比如像本文一样,将坏死性凋亡与ICB治疗抵抗结合,聚焦将“冷”肿瘤变为“热”肿瘤;
2.要想将机制研究转化为临床应用,需要更多考虑机制特异性,药物可行性和是否特异性靶向某种细胞。本文中的小分子化合物通过提高Z-DNA激活ZBP1介导的坏死性凋亡,适用于ZBP1表达且高浸润的成纤维细胞,而不影响正常细胞,也不受限于肿瘤细胞是否表达相关分子,可谓是一种巧思。有时靶向微环境中的细胞,比直接靶向病理状态细胞更有效;
3.擅于利用公共数据库资源。尽管本文中包含大量的测序内容,但在最后还是运用了TCGA和GEO的肿瘤普通转录组和单细胞转录组测序数据,这也启发我们,有的时候公共数据库并一定用来筛选主角分子,也可以用于筛选特定靶向的细胞类群。
只有深入了解机制和分子背景,根据机制巧妙设计解决方案,再结合公共或自测数据进行证实,才能做出摒弃套路的基础研究并进行临床转化。
延伸阅读:其实2021年Cell Reports就发表过一篇关于ADAR1通过抑制ZBP1介导的免疫应答和泛凋亡促进肿瘤发生的文章。这篇文章和本文的差异在哪里?欢迎有兴趣的读者来聊一聊。
文章来源:小张聊科研