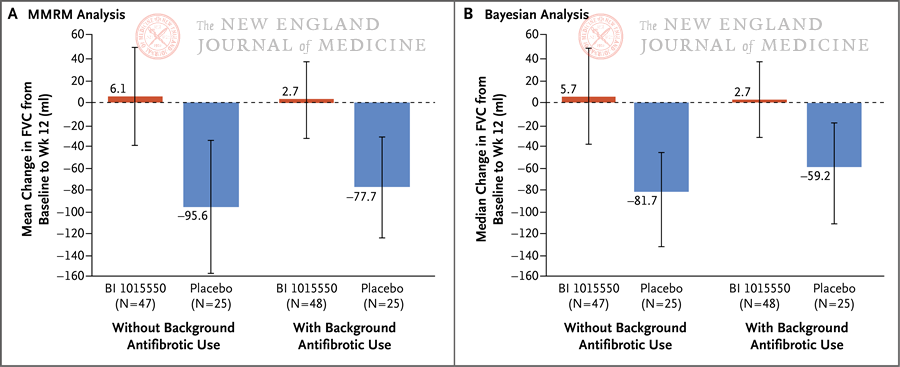
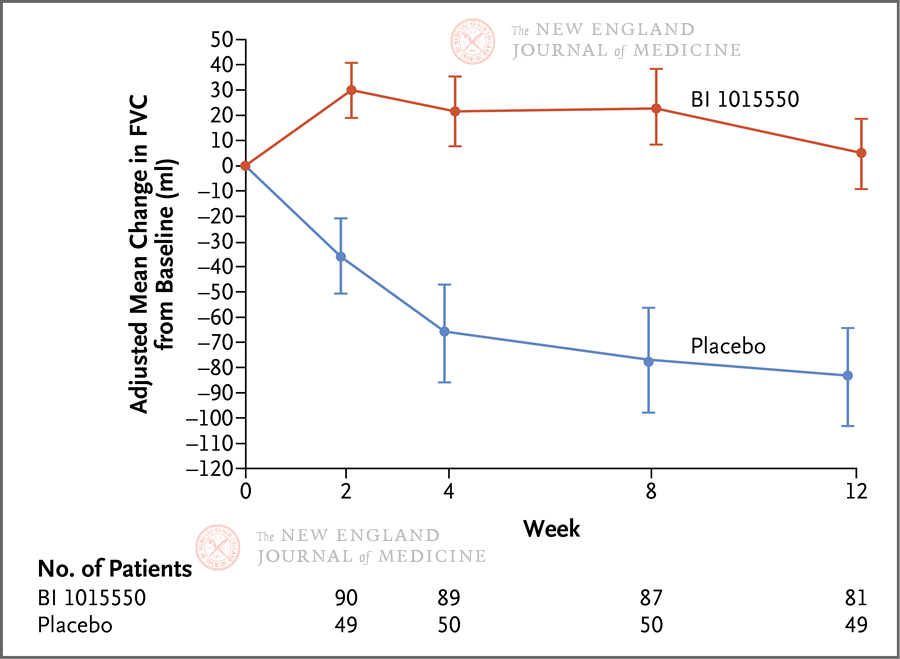
特发性肺纤维化(IPF)是一种病因不明、慢性进行性少见间质性肺疾病,患者死亡率高、生存期短。尼达尼布和吡非尼酮获批是抗纤维化药物开发的高光时刻,但其只能减缓肺功能下降,无法阻止疾病进展。2022年6月9日,《新英格兰医学杂志》(NEJM)发表国际多中心、2期、双盲、安慰剂对照试验。该研究在设计中引入贝叶斯分析,将前期临床试验数据作为历史对照,随机分配更多患者到活性药治疗组,有效利用了相对稀缺的患者资源。其结果表明,磷酸二酯酶4B亚型选择性抑制剂BI 1015550单独用药或与其他抗纤维化药物(尼达尼布或吡非尼酮)联用,在12周治疗期间阻止了特发性肺纤维化患者肺功能下降。詹曦首都医科大学附属北京朝阳医院呼吸与危重症医学科;北京市呼吸疾病研究所特发性肺纤维化(IPF)是一种慢性、进行性、纤维化型间质性肺疾病,发病人群以有吸烟史的中老年人为主,临床上表现为咳嗽、活动后气短,肺功能呈现进行性下降,诊断后中位生存期为2~3年左右[1],甚至低于部分恶性肿瘤,肺移植是唯一能提高预期寿命的治疗方法。IPF的发病率为0.09~0.93/10,000人-年,虽然其在各国有所差异[2],但全球发病率和患病率都在逐年上升[3],且目前尚无特效药物。全球获批的两种抗肺纤维化药物——吡非尼酮[4]和尼达尼布[5],都只能延缓肺功能下降(从无治疗的用力肺活量[FVC]年下降200 mL以上延缓为FVC年下降100 mL左右),不能改善生存率,对于缓解症状和提高生活质量的证据也极其有限[6]。IPF的发病机制尚不明确。目前认为,其发病与早期肺泡上皮细胞的反复损伤和异常修复有关,此外,多种细胞因子、信号通路、遗传因素也在IPF的发生发展中起重要作用。IPF的危险因素包括吸烟、病毒感染、环境污染、慢性误吸、遗传易感性和药物。在肺脏持续进展性纤维化过程中,肌成纤维细胞是主要效应细胞,具有分泌多种炎性细胞因子、促进细胞外基质沉积及肺泡结构破坏的功能。上皮-间充质细胞转化、成纤维细胞增殖、周细胞和循环纤维细胞的介入均促进肌成纤维细胞的分化和增殖。第二信使环磷酸腺苷(cAMP)在IPF的形成过程中抑制成纤维细胞增殖或向肌成纤维细胞分化[7,8]。细胞核中延长的ERK信号传导驱动人肺成纤维细胞的增殖,以响应诸如血小板衍生生长因子(PDGF)之类的生长因子,细胞核中cAMP的持续升高可有效抑制PDGF诱导的核ERK级联反应和成纤维细胞增殖[7]。磷酸二酯酶4(PDE4)是肺成纤维细胞中主要的cAMP降解酶,其水平在纤维化过程中上调,广泛参与炎症过程,在IPF的病理生理过程中也很活跃[9,10]。PDE4的同功酶具有多样性,分4种亚型:PDE4A、4B、4C和4D。前期试验提示选择性的PDE4抑制剂罗氟司特在IPF动物模型的体外和体内均有抗纤维化的功效[11,12]。BI 1015550是PDE4B抑制剂,其治疗特发性肺纤维化的2期临床试验发表于2022年6月9日出版的《新英格兰医学杂志》(NEJM)[13],其结果令人鼓舞(5月15日在线发表)。此临床研究为一项双盲、安慰剂对照试验,共147例患者入组,按2:1的比例随机接受BI 1015550 18 mg每日两次或安慰剂治疗,为期12周,共132名患者完成了试验。主要研究终点为第12周时用力肺活量(FVC)相较于基线值的变化情况。此研究并未排除正在服用其他抗纤维化药物(吡非尼酮或者尼达尼布)的患者,而是根据是否使用抗纤维化药物(定义为在筛选时已经稳定使用至少8周)分为亚组。主要研究结局采用贝叶斯分析。在未服用已批准抗纤维化药物治疗的患者中,BI 1015550试验组患者FVC中位值增加了5.7 mL(95%可信区间,−39.1~50.5),安慰剂组FVC中位值下降了81.7 mL(95%可信区间,−133.5~−44.8),中位差异88.4 mL(95%可信区间,29.5~154.2),BI 1015550优于安慰剂的概率为99.8%。在已服用抗纤维化药物治疗的患者中,试验组患者FVC中位值增加了2.7 mL(95%可信区间,−32.8~38.2),安慰剂组患者FVC中位值下降59.2 mL(95%可信区间,−111.8~−17.9),中位差异69.4 mL(95%可信区间6.3~125.5),BI 1015550优于安慰剂的概率为98.6%。由于此研究为探索性试验,未对多重性进行验证性测试或调整,因此没有报告P值,而是使用了95%可信区间或置信区间(图1和图2)。 图1. 12周时试验组和对照组患者的FVC较基线值的变化,根据是否已经使用抗纤维化药物分为亚组。图2.所有患者的FVC随时间的变化,使用MMRM分析;竖线代表标准误。研究的次要终点是试验开始至治疗结束后1周内发生不良事件的患者比例。最常见的不良事件是腹泻(>10%),其余为胀气、头痛、咳嗽、乏力等,共有13名患者因不良事件提前终止药物试验,其中3名患者因腹泻而退出,但绝大多数腹泻症状轻微。无论是否使用其他抗纤维化药物,试验组的不良事件均多于安慰剂组。试验组2例患者发生了致死性不良事件,1例为新冠肺炎,1例为疑似IPF急性加重。未报告药物相关的抑郁或自杀,但有1例患者在BI 1015550治疗结束后9天时有自杀想法。其余的研究终点还包括治疗12周时患者的生活质量评分较基线值的变化,使用肺纤维化问卷(Living with Pulmonary Fibrosis Questionnaire,L-PF问卷)进行评分。此研究的亮点,除了临床试验允许合并使用抗纤维化药物,值得一提的是研究设计。由于IPF为罕见病,大量入组患者较为困难,因此其对照组患者数据来自尼达尼布2~4期临床药物试验的对照组,使用贝叶斯分析使其信息匹配;为了对抗先验数据冲突,使用了元分析预测(meta-analytic predictive,MAP)优先以确保稳健性。而主要研究终点则通过受限的最大似然法,以重复测量的混合模型(MMRM)进行第一步分析,在此模型的基础上计算试验组和对照组FVC相较于基线值的调整平均变化。受限于较短的研究时间和相对较小的样本量,此临床试验未能收集到有意义的重要临床事件,如急性加重和死亡,以及患者生活质量的变化,这些事件需要3期临床试验来进一步评估。但此研究显示,无论是否已经开始抗纤维化治疗,BI 1015550都能阻止IPF患者FVC的下降,其安全性也能保证进一步研究的开展,前景仍然是光明的、可期待的。参考文献1. Raghu G, Collard HR, Egan JJ, et al. An official ats/ers/jrs/alat statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824.2. Maher TM, Bendstrup E, Dron L, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res 2021;22:197.3. Gao J, Kalafatis D, Carlson L, et al. Baseline characteristics and survival of patients of idiopathic pulmonary fibrosis: A longitudinal analysis of the swedish ipf registry. Respir Res 2021;22:40.4. King TE, Jr., Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083-2092.5. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071-2082.6. Raghu G, Rochwerg B, Zhang Y, et al. An official ats/ers/jrs/alat clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015;192:e3-19.7. Roberts MJ, May LT, Keen AC, et al. Inhibition of the proliferation of human lung fibroblasts by prostacyclin receptor agonists is linked to a sustained camp signal in the nucleus. Front Pharmacol 2021;12:669227.8. Lambers C, Boehm PM, Karabacak Y, et al. Combined activation of guanylate cyclase and cyclic amp in lung fibroblasts as a novel therapeutic concept for lung fibrosis. Biomed Res Int 2019;2019:1345402.9. Li H, Zuo J, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol 2018;9:1048.10. Heukels P, Moor CC, von der Thusen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in ipf pathogenesis and treatment. Respir Med 2019;147:79-91.11. Sisson TH, Christensen PJ, Muraki Y, et al. Phosphodiesterase 4 inhibition reduces lung fibrosis following targeted type ii alveolar epithelial cell injury. Physiol Rep 2018;6:e13753.12. Matsuhira T, Nishiyama O, Tabata Y, et al. A novel phosphodiesterase 4 inhibitor, aa6216, reduces macrophage activity and fibrosis in the lung. Eur J Pharmacol 2020;885:173508.13. Richeldi L, Azuma A, Cottin V, et al. Trial of a preferential phosphodiesterase 4b inhibitor for idiopathic pulmonary fibrosis. N Engl J Med 2022 May 15 (Epub ahead of print).文章来源:NEJM医学前沿